Chapter 15 Saquinavir
INTRODUCTION
On the 4 December 1997, the Society for Medicines Research Award for Drug Discovery was presented to Redshaw, Duncan, and Roberts for their contributions to the discovery of the HIV protease inhibitor (PI) saquinavir. Mapping of the HIV genome had revealed the existence of the virus-specified protease and this had become a target for several of the leading pharmaceutical companies.1 A series of compounds which inhibited the protease were prepared, resulting in the synthesis of Ro 31-8959 (saquinavir) also known as compound XVI.2 Antiviral activity was in the low nanomolar range and this, combined with a relatively clean preclinical safety profile resulted in the development of saquinavir by Hoffman–La Roche (patent #5196438). In 1995, saquinavir became the first HIV PI approved by the US Food and Drug Administration (FDA).3 It was produced as a hard gel formulation (saquinavir-HGC), known as Invirase.
Saquinavir-HGC had poor oral bioavailability and so two further formulations were developed. In 1997, in the era when PIs were administered without ritonavir boosting, a soft gel capsule (saquinavir-SGC) marketed as Fortovase was approved for use. At the recommended dosage of 1200 mg three times daily (tid) it provided much higher plasma saquinavir concentrations and drug exposure than those attained with the conventional saquinavir-HGC formulation, administered at the recommended dosage of 600 mg tid.4,5 Unfortunately, saquinavir-SGC was associated with an increase in gastrointestinal side effects.6 It contained the excipient capmul which is absent in the saquinavir-HGC formulation which was probably responsible for the gastrointestinal problems seen with the saquinavir-SGC formulation. The large size of the saquinavir-SGC capsules and their need for refrigeration was also a problem. A few years later, however, low-dose ritonavir was demonstrated to significantly improve the pharmacokinetic profile of saquinavir-HGC. Saquinavir-SGC was no longer needed and production was discontinued in February 2006. In addition, the reformulation of saquinavir-HGC into a 500 mg tablet as saquinavir-500 has reduced the daily pill burden from ten tablets to four which has obvious adherence advantages. Saquinavir-500 is the only formulation of saquinavir being marketed, although saquinavir-HGC is still available for the few patients who prefer to remain on it.
CHEMICAL FORMULATION
Saquinavir is a peptide-like, hydroxyethylamine transition state analogue of the human immunodeficiency virus type 1 (HIV-l) protease. The chemical name for saquinavir mesylate is N-tert-butyl-decahydro-2-[2 (R)-hydroxy-4-phenyl-3 (S)-[[N-(2 quinolylcarbonyl)-L-asparaginyl]amino]butyl]-(4aS,8aS)-isoquinoline-3 (S)-carboxamide methanesulfonate with a molecular formula C38H50N6O5. CH4O3S and a molecular weight of 766.96Da.7 The molecular weight of the free base is 670.86. The structural formula of saquinavir mesylate can be seen in Figure 15-1.
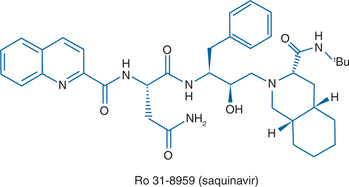
Saquinavir mesylate is a white to off-white, very fine powder with an aqueous solubility of 2.22 mg/mL at 25°C. Saquinavir-500 as the mesylate is available as a light orange to grayish- or brownish-orange, oval cylindrical, biconvex film-coated tablet for oral administration in a 500 mg strength (as saquinavir free base). Each tablet also contains the inactive ingredients lactose, microcrystalline cellulose, povidone K30, croscarmellose sodium, and magnesium stearate. Each film coat contains hypromellose, titanium dioxide, talc, iron oxide yellow, iron oxide red, and triacetin.
IN VITRO ANTIVIRAL ACTIVITY
Saquinavir is a potent in vitro inhibitor of the proteases of both HIV-l and HIV-2, with a Ki of 0.1 nM.2,8 It has virtually no activity against various mammalian proteases, such as pepsin and cathepsin, with Ki values higher than 10 000 nM.1 In vitro using the outcome measures of inhibition of HIV-l p24 antigen production, inhibition of syncytium formation, or cell viability assays.1–3,9,10
Saquinavir has potent (nanomolar) anti-HIV activity against a wide variety of laboratory and clinical isolates, including: zidovudine-sensitive and zidovudine-resistant isolates; in acute and chronic infection systems; in infected T-lymphocyte lines, monocytic cell lines, and peripheral bloodmononuclear cells (PBMCs). For example, in T-lymphocyte lines the range of 50% inhibitory concentration (IC50) values was 0.5–7.0 nM, and the range of 90% inhibitory concentration (IC90) values was 6–34 ηM. In PBMCs the IC50 values range from 3.5 to 10.0 nM. However, specific IC50 and IC90 values from in vitro experiments may not directly reflect the in vivo activity of saquinavir because of factors such as plasma protein binding and intracellular accumulation of drug.5
SAQUINAVIR IN COMBINATION WITH OTHER AGENTS
In a range of cell lines, saquinavir in combination with various other antiretroviral drugs, (compared with the activity of saquinavir alone) showed additive or synergistic activity against various strains of HIV-l without increased cytotoxicity.11–13 Although saquinavir and indinavir showed antagonistic activity in vitro against a range of clinical isolates of HIV-l at all drug concentrations investigated,14 recent data suggest that saquinavir is highly synergistic with other PIs (Table 15-1).
Table 15-1 Synergistic Anti-HIV Activity of Saquinavir with FDA Approved HIV-1 Agents Determined in Drug Combination Studies15
| Saquinavir + | Mean Synergy/Antagonism Volume (μM2%; n = 3) | Interpretation < −50 μM2 % = antagonistic -50 to 50 μM2% = Additive 550–100 μM2% = Synergistic >100 μM2%= Highly synergistic |
|---|---|---|
| Amprenavir | 473/−4.5 | Highly synergistic |
| Atazanavir | 205/−0.5 | Highly synergistic |
| Indinavir | 151/−8.1 | Highly synergistic |
| Lopinavir | 224/−31 | Highly synergistic |
| Nelfinavir | 275/−8.3 | Highly synergistic |
| Ritonavir | 237/−6.9 | Highly synergistic |
CYTOTOXICITY
Saquinavir has a high therapeutic index in vitro and only shows cytotoxicity at concentrations more than 1000 times higher than those producing inhibition of HIV-1.16 In CD4 T lymphocyte cell lines, concentrations of saquinavir producing 50% toxicity (assessed by cell viability markers) ranged from 5 to 100 nmol/L. The in vitro effects of various concentrations of saquinavir on the development of mature virions has been assessed and has demonstrated decreasing amounts of mature particles with increasing concentrations of saquinavir from 10 nM up to 1000 nM concentrations of saquinavir. At the highest concentrations none of the virions produced had a mature appearance.17
In a model of chronic infection in MT-4 cells with an HIV-1 laboratory strain, 100 nM of saquinavir completely suppressed evidence of infection for 87 days. HIV-l reemerged in this culture system when saquinavir was removed from the culture medium after a minimum of 11 days postinfection.18
PHARMACOKINETIC PROPERTIES
Absorption and Distribution
After administration of single 1200 mg intravenous doses of saquinavir to eight adult volunteers, the mean steady-state volume of distribution (Vd) was 700 L, suggesting that saquinavir is widely distributed in body tissues. However, concentrations of saquinavir in the cerebrospinal fluid (CSF) appear to be negligible. In 11 HIV-infected patients treated with saquinavir-HGC plus ritonavir as part of a combination therapy, a CSF:plasma saquinavir concentration ratio of ≤0.005 (0.5%) was observed in all study subjects.19
Protein Binding
Saquinavir is 97% bound to serum proteins, and is similar to the degree of protein binding reported with lopinavir (98–99%), nelfinavir (98%), amprenavir (90%), and indinavir (90%). Binding is predominantly to alpha-1-acid glycoprotein. Saquinavir is also a high-affinity substrate for the drug transporting protein P-glycoprotein (Pgp). Pgp binding is thought to contribute to the limited oral bioavailability of the drug.20
Elimination
Elimination of saquinavir is predominantly nonrenal, with ∼1% of the drug excreted unchanged in the urine and 88% of the nonboosted dose eliminated in the feces as unchanged drug and metabolites. Several mono- and dihydroxylated metabolites are produced, which have no significant antiviral activity.21
As biliary excretion appears to be the major route of elimination, the pharmacokinetics of the drug are not expected to be markedly affected in patients with renal impairment5 but no definitive data are available.
Metabolism
There is first-pass metabolism, predominantly by the cytochrome P450 3A4 (CYP3A4) isoenzyme in the liver and small intestine.22,23 Drugs that induce the CYP3A4 isoenzyme such as rifampicin (rifampin), rifabutin, phenytoin and carbamazepine have the potential to decrease concentrations of saquinavir. Conversely, administration of saquinavir with drugs that inhibit CYP3A4, such as the PI ritonavir, results in an increase in the concentrations of saquinavir formulations.24
Bioavailability
In healthy volunteers the mean absolute bioavailability of saquinavir after a single oral 600 mg dose of the HGC formulation taken with food is only 4%. This is because of both limited absorption and extensive first-pass metabolism. The steady-state saquinavir exposure appears to be greater in HIV-infected people than in healthy volunteers.25
For historical interest only it was shown that the oral bioavailability of saquinavir-SGC was three to four times higher than that of the saquinavir-HGC formulation and the area under the plasma concentration–time curve (AUC) was more than eight times higher in recipients of saquinavir-SGC l200 mg tid than after saquinavir-HGC 600 mg tid. Again, the absorption profile of saquinavir-SGC was different between healthy volunteers and patients with HIV-1 infection, with mean AUC and Cmax values in HIV-infected patients being approximately twice those in healthy volunteers.5,26
Food Interactions
As was observed in pharmacokinetic investigations of the saquinavir-HGC formulation, absorption of saquinavir-500 is markedly enhanced by the presence of food especially if its fat content is high. Twenty-two HIV-infected adults were given a high fat meal (46-66 g of fat, 1070–1090 kcal) versus fasted state with three consecutive doses of 1000 mg saquinavir-500 with ritonavir 100 mg. Saquinavir PK parameters were ∼70% reduced during administration under fasting conditions compared with administration following the high-fat, high-calorie meal. There was no significant change in ritonavir pharmacokinetics. The food effect seen with saquinavir-500/ritonavir in this study was less than that previously seen with saquinavir-HGC without ritonavir. Interestingly, the majority of subjects (2½2) maintained plasma saquinavir levels above the therapeutic threshold (100 ng/mL) even in the fasted state. However, saquinavir-500 and ritonavir should still be administered with food to ensure maximum therapeutic value.27
PHARMACOKINETICS OF SAQUINAVIR BOOSTING BY LOW-DOSE RITONAVIR
Saquinavir was one of the first antiretroviral agents for which an increase in plasma drug levels was demonstrated when given in combination with ritonavir. This was later found to be related to profound inhibition of CYP450 pathway responsible for metabolism of saquinavir and other antiretroviral medications by ritonavir. The dose regimen initially used was 400/400 mg bid saquinavir-SGC or saquinavir-HGC/ritonavir. The significant increase of saquinavir levels in the presence of ritonavir was demonstrated in an open-label, randomized, parallel, multiple-dose study involving 64 healthy HIV-1-negative volunteers who were given saquinavir-HGC (800 mg bid) alone, ritonavir (400 mg bid) alone, or a combination of saquinavir (400–800 mg bid) with ritonavir (200–400 mg bid) for 14 days.28 Among those given saquinavir at a dose of 800 mg bid, there were overall 17-, 22-, and 23-fold increases in saquinavir (AUC-24 h) when ritonavir was administered at doses of 200, 300, and 400 mg bid, respectively.
It was later found that lower doses of ritonavir are sufficient to ‘boost’ saquinavir levels29–31 and saquinavir/ritonavir is now frequently prescribed using 1000 mg of saquinavir with 100 mg of ritonavir twice daily (bid) or 2000 mg of saquinavir with 100 mg of ritonavir once daily (qd). The ritonavir should be given simultaneously in order to maximally increase saquinavir exposure.32 Pharmacokinetics of the currently recommended dose was investigated in 24 healthy volunteers in a study that compared saquinavir-SGC/ritonavir (1000/100 mg bid) with saquinavir-HGC/ritonavir (1000/100 mg) in which each subject was given each combination separately for 10 days. Saquinavir-HGC/ritonavir led to significantly higher plasma saquinavir levels than saquinavir-SGC/ritonavir for all the pharmacokinetic variables evaluated.33 Addition of ritonavir leads to significant increases in saquinavir exposure irrespective of whether the saquinavir-HGC or saquinavir-500 formulation is used.31,34–37
Increasing the dose of ritonavir above 100 mg does not improve the pharmacokinetic profile of saquinavir. A combined pharmacokinetic analysis of two dose-ranging studies indicates that the boosting effect on saquinavir is similar over the dose range 100–400 mg bid of ritonavir.30 Furthermore, the combination of saquinavir/ritonavir 1000 mg/100 mg bid resulted in higher exposures to saquinavir in HIV-1 infected patients compared with the exposure to saquinavir seen historically using a saquinavir/ritonavir 400 mg/400 mg bid combination.38 Since the toxicity of ritonavir is directly proportional to its plasma levels39–41 use of low ritonavir doses 100–200 mg/day diminishes the potential for ritonavir-associated adverse effects such as nausea, vomiting, oral paraesthesia, taste perversion and elevated lipid levels.40 Saquinavir does not alter the pharmacokinetic parameters of ritonavir.29
ONCE-DAILY PHARMACOKINETICS
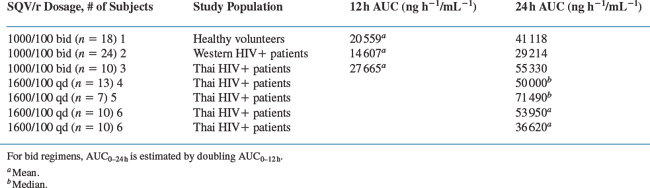
Although bid dosing is currently used in clinical practice for combination saquinavir/ritonavir regimens, once-daily (qd) dosing might be a viable alternative42 A comparison of the saquinavir exposures using the AUC for different doses and dosing regimens can be seen in Table 15-2.
PHARMACOKINETICS OF SAQUINAVIR IN CHILDREN
The clinical pharmacologic characteristics of saquinavir-SGC have been investigated in HIV-1-infected children and adolescents. It was shown that the pharmacokinetics of saquinavir in children is different from that of adults. However, the study only looked at unboosted saquinavir so the relevance to today’s practice is limited. PK was assessed after single-dose administration and after short- and long-term administration of 50 mg/kg saquinavir-SGC tid. The single-dose pharmacokinetics of fixed (1200 mg) versus unrestricted weight-adjusted dosing (50 mg/kg) was also investigated. Saquinavir-SGC in children resulted in lower saquinavir exposure (steady-state geometric mean AUC from time 0–24 h, 5790 ng h−1 mL−1; steady-state concentration 8 h after drug administration (C (8 h, SS)), 65 ng/mL) and adolescents (steady-state geometric mean AUC (0–24 h), 5914 ng h−1 mL−1) than that reported in adults treated with 1200 mg tid (steady-state geometric mean AUC (0–24 h), 21 700 ng h−1 mL−1; C (8 h, SS), 223 ng/mL).43
SAQUINAVIR IN PREGNANCY
Saquinavir is in FDA pregnancy category B with respect to teratogenicity based on the available preclinical data. Saquinavir was not mutagenic in vitro in the Ames test in bacteria or in mammalian cells, and it did not induce chromosomal damage in vitro in PBMCs or in vivo in the mouse. An Antiretroviral Pregnancy Registry has been established to monitor the outcomes of pregnant women exposed to antiretroviral agents, including saquinavir (http://www.apregistry.com). It is not known whether saquinavir crosses the placenta in humans or whether it is secreted in human breast milk. It is secreted in the milk of laboratory rats.44 There are few studies in pregnant women but there are observational data on the use of boosted saquinavir in pregnancy.
In a study of the new 500 mg saquinavir formulation at a dose of 1000 mg saquinavir with 100 mg ritonavir bd, there was no significant difference in saquinavir PK between the second and third trimester of pregnancy. This is in contrast to observations with other PIs such as nelfinavir, lopinavir and ritonavir.44a
LOW-DOSE BOOSTED SAQUINAVIR TWICE-DAILY (BID) IN PREGNANCY
From December 2002 to July 2005, 13 women with a total of 15 pregnancies received saquinavir-HGC with ritonavir (1000/100 mg bid) plus Combivir from a median of 21 weeks (range 13–25 weeks) until delivery. The median age of the women was 32 years (range 19–39 years); the median CD4 T-lymphocyte count at initiation of therapy was 437 cells/μL (range 237–637 cells/μL) and viral load 7901 copies/mL (range 117–55 460 copies/mL). Seven women had received HAART previously in pregnancy. One woman was switched from nevirapine/Combivir due to nevirapine-associated hepatic toxicity. All women had a HIV RNA level of <50 copies/mL at delivery. The median period of gestation was 37 weeks and 73% of women had a caesarean delivery. Sixteen HIV-negative infants including one set of twins were born during the study period. No additional resistance mutations from baseline were detected on stopping therapy post delivery.45
LOW-DOSE BOOSTED SAQUINAVIR ONCE-DAILY (QD) IN PREGNANCY
A cohort of 38 women pregnant women received an initial saquinavir/ritonavir dose of 1200/100 mg qd. Therapeutic drug monitoring (TDM) was used to guide the dosage of the boosted saquinavir containing therapy to maintain a saquinavir Cmin of 100 ng/mL (24 ± 5 h post dose). Their median age was 31 years (range 21–38 years). At inclusion 15 (44.1%) women were drug naive, 33 (86.8%) had no prior PI failure, and 11 (32.3%) were already on treatment. Their median CD4 T-lymphocyte count was 508 cells/μL (range 42–1158 cells/μL) and viral load 3905 copies/mL (range <50–181 000 copies/mL). Therapy was initiated at a median of 20 weeks (range 0–35 weeks) of pregnancy and 30 (78.9%) women received zidovudine and lamivudine as their nucleosides. The saquinavir dose was increased to 1600 mg qd in two women. After a median of 17 weeks (range 3–39 weeks), 29 women had an undetectable viral load at delivery. Three women had viral loads of 110 copies/mL, 400 copies/mL and no data, respectively, but all three had started saquinavir 3–4 weeks before delivery. Mode of delivery was vaginal in 16 pregnancies and 16 by caesarean section (11 of these were for gynecological reasons). The authors reported no mother to child transmissions.46,47
SAQUINAVIR USE AND LIVER DISEASE
Saquinavir is metabolized by the liver, and it should be used with caution in patients with hepatic insufficiency. There are few data investigating its use in patients with chronic liver disease as patients with baseline liver function test results higher than five times the upper limit of normal were excluded from clinical studies. In the MaxCmin1 study 24/306 or 8% of patients had chronic hepatitis. No significant changes were seen in alanine transaminase (ALT) or aspartate transaminase (AST) in the boosted saquinavir or indinavir group after 48 weeks even in those who had abnormal values at baseline.48
DRUG INTERACTIONS
The potential for drug interactions between saquinavir and other drugs is significant because the CYP3A4 isoenzyme is a common metabolic pathway for many agents.22 Drugs that are metabolized by or induce or inhibit the cytochrome P450 system, especially isoenzyme 3A4, may affect the metabolism, plasma levels, and systemic exposure of saquinavir. In addition, saquinavir is a mild inhibitor of cytochrome P450 enzymes and may affect the metabolism of some other drugs. Furthermore, as saquinavir is a substrate for Pglycoprotein (Pgp), drugs that modify Pgp activity may modify the pharmacokinetics of saquinavir. Similarly, saquinavir has the potential to modify the pharmacokinetics of other drugs that are substrates for Pgp.23,49
Antiviral Agents
Concomitant use of certain other antiretroviral agents with saquinavir may significantly increase or decrease saquinavir plasma concentrations. These antiretrovirals include delavirdine (Rescriptor), indinavir (Crixivan), nelfinavir (Viracept), and ritonavir (Norvir).14,23 There is little clinical relevance apart from the ritonavir interaction as these drugs are rarely prescribed together. Saquinavir-HGC has no clinically significant interaction with nevirapine, lopinavir/ritonavir or with fosamprenavir/ritonavir. There is no clinical significant interaction of saquinavir with tenofovir or abacavir.
Statins
Caution should be used when any HIV PIs, especially when given with ritonavir, are used concurrently with HMG-CoA reductase inhibitors that are metabolized by the CYP3A4 pathway. The use of saquinavir with lovastatin or simvastatin is not recommended. Care should be taken when co-administration of atorvastatin, rosuvastatin, or cerivastatin is contemplated and patients should start with a low dose as the drug interaction may lead to raised concentration of statins which can cause myopathy or rhabdomyolysis.50
Rifampicin (Rifampin)
Recent data from a 28-day phase I clinical trial of saquinavir/ritonavir 1000 mg/100 mg bid and rifampin 600 mg qd in normal healthy volunteers showed significant hepatocellular toxicity in ∼40% of patients with transaminase elevations of up to 20 times the upper limit of normal. Following drug discontinuation, clinical symptoms resolved and liver function tests returned to normal.51 Although such an interaction has not been seen in HIV-1 patients who have had co-administration of these drugs it is recommended that rifampin should not be given to patients taking ritonavir-boosted saquinavir as part of combination antiretroviral therapy.
Efavirenz
Saquinavir levels can be significantly reduced when prescribed with efavirenz. In a previous pharmacokinetic study of the saquinavir and efavirenz interaction, it was found that efavirenz reduced the AUC of saquinavir by 62%, while the AUC of efavirenz remained almost unchanged. However, with the addition of low-dose ritonavir the decrease in saquinavir AUC was only 19%.52 This interaction requires further investigation.
Atazanavir
A study of 18 HIV-1 patients receiving saquinavir/ritonavir switched to 1600/100 mg qd a minimum of 3 days before atazanavir (300 mg qd) was added to the regimen. Atazanavir was discontinued on day 32. Saquinavir and ritonavir pharmacokinetic parameters, with and without atazanavir were analyzed and a safety analysis was performed at screening, days 1, 11, 32, and follow-up. The addition of atazanavir to saquinavir/ritonavir increased saquinavir Ctrough, Cmax and AUC0–24 by 112%, 42%, and 60%. Ritonavir Cmax and AUC0–24 increased by 34% and 41%. The regimen was well tolerated, with no significant change in laboratory parameters, except for the occurrence of hyperbilirubinemia. Four patients developed scleral icterus and two jaundice. Total and unconjugated bilirubin increased approximately fivefold during atazanavir therapy.53
In a further study, reduced doses of atazanavir administered at 150 and 200 mg qd, in the presence of low-dose ritonavir were still capable of increasing saquinavir exposure with the lowest doses limiting the occurrence of hyperbilirubinemia without unduly boosting ritonavir concentrations.54
Omeprazole
Proton pump inhibitors such as omeprazole can be bought as an over-the-counter medicine in many countries. The potential for them to cause significant interactions with PIs was seen with both boosted and unboosted atazanavir where significant reductions in all pharmacokinetic parameters. A study of omeprazole 40 mg qd given to 18 healthy volunteers taking saquinavir-500 with ritonavir 1000/100 mg at steady state showed a twofold increase in saquinavir Cmin trough and AUC-12 h plasma exposure. There was no significant increase in ritonavir PK parameters. Despite the increased plasma saquinavir exposure, no clinically significant or grade 3/4 laboratory adverse events were recorded. The exact mechanism of interaction is not known but appears to be due to increased absorption as t½ and time to Cmax were not affected.55
A similar study in 18 HIV positive patients also showed similar increases in saquinavir levels with omeprazole without significant toxicity.55a
Methadone
With ritonavir-boosted saquinavir there was a small reduc-tion in the free fraction of R-methadone.56 However, it is not clinically significant so no methadone dose changes are required.
Oral Contraceptives
There are no data available regarding effects of saquinavir on ethyl estrogen or norethisterone levels. Saquinavir kinetics are not affected by the oral contraceptive pill.57
Herbal Agents
Use of saquinavir with St John’s wort (Hypericum perforatum) or products containing St John’s wort can potentially decrease saquinavir concentrations and lead to loss of virologic response and consequent resistance.58
Piscitelli reported that garlic supplements reduced blood levels of saquinavir by half during a 40-day study in which saquinavir was dosed at 1200 mg on days 1–4, 22–25, and 36–39 and garlic capsules were given from days 5 to 25. After a 10-day washout period from garlic treatment (saquinavir measured at day 36), trough levels of saquinavir had reached 70% of their baseline value.59
Others
Co-administration of certain other drugs with saquinavir may cause an increase or decrease in plasma concentrations of saquinavir or of the co-administered drug. Caution should be used when the following drugs are used concomitantly with saquinavir: calcium channel blockers, carbamazepine, clarithromycin, clindamycin, dapsone, dexamethasone, ketoconazole, phenobarbital, phenytoin, quinidine, rifabutin, and ergot derivatives.5 Saquinavir should not be co-administered with astemizole or cisapride owing to the increased risk of cardiac arrhythmias. Other drugs, including midazolam and triazolam, can lead to prolonged sedation.
TOXICITY
In controlled studies more than 5000 patients have received saquinavir-HGC, and more than 850 patients have received saquinavir-SGC. Overall, saquinavir appears to be well tolerated. Some data are available from early studies in which saquinavir-HGC was administered as monotherapy, but most available data are from the use of saquinavir-HGC or saquinavir-SGC in combination with other antiretroviral agents. The most frequent side effects with both formulations are gastrointestinal and include diarrhea, nausea, and abdominal discomfort or pain.60 Most treatment-related side effects have been mild. Overall, moderate or severe gastrointestinal side effects at least possibly related to treatment have occurred in 5–10% of patients receiving saquinavir-HGC and 10–20% of patients on saquinavir-SGC therapy; additional patients may have milder symptoms. Some of the GI intolerance to saquinavir-SGC was probably related to excipients in the formulation. Other toxicities that occurred in more than a few percent of patients treated included headaches, fatigue, and elevations in serum transaminases. Elevations in creatine phosphokinase have also been observed but are believed to be related to HIV-1 or other therapies, rather than to saquinavir.
Metabolic complications have been increasingly described in patients receiving PI therapy. On the basis of postmarketing surveillance reports, the FDA issued an alert in 1997 about a possible relation between PI use and the development or worsening of diabetes mellitus, hyperglycemia, and diabetic ketoacidosis; some of the reported cases occurred in persons receiving saquinavir.61 There have also been other metabolic complications in persons receiving PIs, including body habitus changes, intraabdominal fat deposition, hypertriglyceridemia, and hypercholesterolemia.62–67 The frequency and relation of these events to treatment with either saquinavir formulation is unknown.
Lipodystrophy
Lipoatrophy is associated with nucleoside analogue treatment especially thymidine analogs. Fat accumulation especially abdominal with an increase in waist size and abnormal waist hip ratio, buffalo hump, and breast enlargement are associated with PI use. In early studies of saquinavir larger doses of ritonavir were used from 400 to 600 mg and the prevalence of fat accumulation as assessed by physical examination was 4–14% at 5 years.68
An assessment of the prevalence and severity of lipodystrophy (LD) after 48 weeks was performed in the MaxCmin1 trial of indinavir/r (IDV/r) 800/100 mg bid or saquinavir/r (SQV/r) 1000/100 mg bid combined nucleosides and non-nucleosides. LD Case definition study questionnaires for patients and physicians were used at week 48. Fat accumulation severity scores were significantly higher in females (P < 0.001 for both), with no difference in lipoatrophy severity score (p = 0.79).69
The contribution of ritonavir to these metabolic and morphological changes was examined in a randomized pilot study evaluating the safety, tolerability, and efficacy of qd atazanavir plus saquinavir, as compared with bid ritonavir plus saquinavir, both in combination with two NRTIs.70 Small lipid changes from baseline with atazanavir/saquinavir were not clinically significant in comparison with the prompt, marked and sustained changes of a magnitude that suggests clinical relevance that occurred in the ritonavir/saquinavir group.



Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


