Fig. 7.1
Schematic representation of coupling between osteoblast, osteocyte, and osteoclast development and function. MSC mesenchymal stem cells, HSC hematopoietic stem cells
Three types of cells are involved in bone remodeling: osteoclasts which resorb an old or damaged bone, osteoblasts which form new bone at the site of the resorbed cavity, and osteocytes which orchestrate the whole process. Osteoclasts and osteoblasts/osteocytes develop from two distinct populations of stem cells residing in the bone marrow, hematopoietic stem cells (HSC) and mesenchymal stem cells (MSC), respectively. Osteoclast differentiation is determined by both, factors produced by cells of osteoclast lineage and factors produced by other bone marrow cells including cells of osteoblast lineage [2]. Osteoclast recruitment from the HSC pool and their maturation is controlled by osteoblast-derived cytokines: M-CSF, IL-6, and RANKL. Osteoblasts originate in a marrow MSC compartment which also produces adipocytes [3, 4]. The commitment of MSC toward either the osteoblast or adipocyte lineage occurs by a stochastic mechanism [5]; lineage-specific transcription factors, such as Runx2, Dlx5, and Osterix for osteoblasts and PPARγ2 and C/EBPs for adipocytes are activated [6–11]. Activation of osteoblast-specific transcription factors is determined by a milieu of extracellular factors, which regulate the cellular activity of Wnt, TGFβ/BMP, and IGF-1 signaling pathways [12]. Process of bone remodeling is controlled by osteocytes, which represent specialized cells of osteoblast lineage [13]. They are located inside of bone mineralized matrix and communicate with other osteocytes and bone marrow environment through the system of dendrite-like processes. Osteocytes control dynamics of bone remodeling process by secreting RANKL to control bone resorption and sclerostin to control bone formation [13].
Bone as an Integral Part of Energy Metabolism System
Bone is closely integrated with the system regulating energy balance. Organs involved in this regulation including brain, fat, gastrointestinal system, and pancreas are secreting hormones which in endocrine manner regulate both energy metabolism and bone mass. Their effect on bone is possible because bone marrow cells, both mesenchymal and hematopoietic lineage, are equipped with necessary receptors to respond to these signaling (Fig. 7.2).
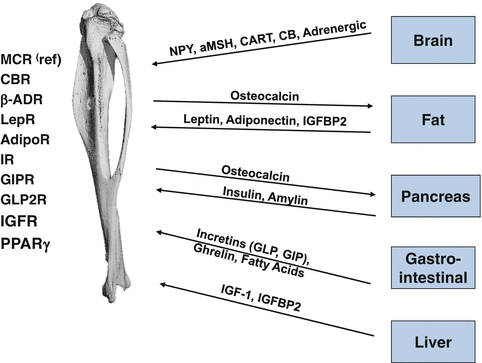
Fig. 7.2
Bone is a part of energy metabolism network. Factors and their receptors which mediate a cross-talk between bone and other organs involved in regulation of energy metabolism. MCR melanocortin receptor, CBR cannabinoid receptor, LepR leptin receptor, AdipoR adiponectin receptor, GIPR glucose inhibitory protein receptor, GLP2R glucagon inhibitory peptide receptor, β-ADR beta adrenergic receptor, IR insulin receptor, IGFR insulin growth factor receptor, PPARγ peroxisome proliferator activator receptor γ
Integration of bone metabolism with energy metabolism has been presented recently as a model which links anabolic effect of insulin signaling in osteoblasts with bone turnover and regulation of insulin sensitivity in peripheral organs [14, 15]. Thus, in osteoblasts insulin signaling regulates an expression of Runx2 and osteocalcin production. In addition, insulin increases support for osteoclastogenesis by decreasing an expression of OPG, a decoy receptor for RANKL. As a result, insulin increases bone turnover and production of undercarboxylated osteocalcin, which in endocrine fashion regulates insulin release from β-cells in pancreas and production of adiponectin in fat tissue [14–17]. Although it is not clear whether this regulatory circuit is affected in diabetes, several studies suggest that patients with T2DM have decreased bone turnover [18–20]. If so, it would result in the decrease in osteocalcin production, especially its undercarboxylated form, which would lead to the attenuation of signaling responsible for increasing of insulin release from the pancreas and increasing fat sensitivity to insulin.
Anti-hyperglycemic Therapies and Their Effects on Bone
The most common form of diabetes is insulin-independent T2DM, which is characterized by insulin and glucose intolerance, and is associated with development of hyperglycemia and hyperinsulinemia. Therapies, either approved by FDA or in Phase III clinical trial, include insulin sensitizers, insulin secretagogues, and drugs which increase glucose excretion in the urine (SGLT2 inhibitors), regulate glucose absorption in intestine (amylin analog), and prevent digestion of carbohydrates (Alpha-glucosidase inhibitors) (Table 7.1).
Table 7.1
Antidiabetic drugs and their effects on skeleton
Target | Mode of action | Class of drugs | Drugs | Skeletal effect |
|---|---|---|---|---|
Insulin | Sensitizers | Biguanides | Metformina | Neutral |
TZDs (PPARγ agonists) | Pioglitazoneb, Rosiglitazoneb | Bone loss; increased fractures | ||
Dual PPARα/PPARγ agonists | Aleglitazarc | Unknown | ||
Secretagogues | K+ ATP | Sulfonylureas (e.g. Glyburidea) | Neutral | |
Meglitinides (e.g. Nateglinide) | Unknown | |||
GLP-1 analogs | Exenatide, Liraglutide, Taspoglutidec, Albiglutidec, Lixisenatidec | Unknown | ||
DPP-4 inhibitors | Alogliptinc, Saxagliptin, Sitagliptin, Vildagliptin, Linagliptin | Decreased fractures | ||
Analogs/other insulinsa | Insulin lispro, Insulin aspart, Insulin glargine | Increased fractures | ||
Other | SGLT2 inhibitors | Canagliflozin, Dapagliflozin | Increased fractures | |
Amylin analog | Pramlintide | Unknown | ||
Alpha-glucosidase inhibitors | Acarbose, Miglitol, Voglibose | Unknown | ||
In general, there is a lack of rigorous clinical evidence regarding the skeletal effects of these medications. Fracture is the primary outcome of interest but is a relatively rare outcome, requiring large studies. Randomized controlled trials (RCT) of diabetes medications have not included fracture as a primary endpoint, but increasingly studies are reporting the evidence from fractures identified as adverse events in RCTs. This provides the best evidence available to us regarding the clinical effects of these medications on the skeleton. Large observational studies have also considered the effects of diabetes medications on fracture risk. However, medications may be systematically prescribed to patients with different risk profiles for fracture, making it difficult to distinguish effects of the medications themselves on fracture, a problem known as “allocation bias” or “confounding by indication.” Changes in bone mineral density are an important marker for skeletal health, but are not always a consistent predictor of the effects of a medication on fracture risk [21]. In addition, diabetic bone is compromised by other deficits, distinct from BMD, that have not been clearly delineated but may include increased cortical porosity and greater accumulation of advanced glycation endproducts in bone collagen (see Chap. 9). As research clarifies which secondary markers are important predictors of fracture risk in diabetic patients, it will be essential to clarify how they are affected by diabetic medications.
Biguanides (Metformin)
Metformin is the most commonly used to increase insulin sensitivity in diabetic patients. Biguanides class of drugs decreases hepatic glucose production and increases glucose uptake in muscle. Metformin is considered by the World Health Organization an essential medicine satisfying the criteria of the public health relevance, evidence on efficacy and safety, and comparative cost effectiveness (www.who.int/medicines). Metformin mechanism of insulin sensitization includes activation of hepatic and muscle AMP-activated protein kinase (AMPK), which results in suppression of fatty acid synthesis, stimulation of fatty acid oxidation in liver and increase in muscle glucose uptake [22]. AMPK also decreases expression of sterol-regulatory element-binding-protein 1 (SREBP-1), a transcription factor involved in adipocyte differentiation and pathogenesis of insulin resistance, dislipidemia and diabetes. Animal studies indicate that metformin has a positive effect on osteoblast differentiation due to increased activity of osteoblast-specific Runx2 transcription factor via AMPK/USF-1/SHP regulatory cascade [23] and it has a negative effect on osteoclast differentiation and bone loss after ovariectomy by decreasing RANKL and increasing osteoprotegerin levels [24]. Interestingly, in rodent models metformin can prevent the adverse effects of TZDs on bone by either inducing reossification of bone after rosiglitazone treatment or preventing rosiglitazone effects when applied in combination with rosiglitazone [25].
Human studies of the effects of metformin on the skeleton are limited in design and number. The only RCT with a fracture outcome that included metformin is the ADOPT trial, discussed in more detail in the section on “Thiazolidinediones (Rosiglitazone, Pioglitazone)” [26]. Briefly, this trial randomized participants to receive metformin, glyburide (a sulfonylurea), or rosiglitazone; the primary outcome was time to monotherapy failure. Fractures were identified as adverse events. The fracture rates were similar in those randomized to metformin or glyburide. During the first 12 months of ADOPT, changes in the levels of the bone resorption marker CTX were similar in women (difference in 12-months change: +2.0 %) and modestly greater in men (−8.4 %) in those assigned to metformin compared with glyburide [27]. The metformin group had greater decreases in levels of the bone formation marker P1NP (difference in 12-months change: −9.4 % women; −19.5 % men), compared with glyburide.
Several observational studies have reported a lower risk of fracture with metformin use. In a study of the Danish population, metformin use was associated with lower fracture risk, compared with nondiabetic residents [28]. In the Rochester cohort, metformin use was associated with a lower rate of fracture in T2DM patients (adjusted hazard ratio 0.7; 95% CI 0.6–0.96) [29]. However, other studies have found no difference in fracture risk with metformin use [30–35]. Interestingly, results from a study of hip fracture in Scotland suggest that metformin tends to be prescribed to patients with a lower overall risk of fracture while sulfonylureas are prescribed to those at higher risk [33]. This finding suggests that some of the reduction in fracture risk reported with metformin use in observational studies may be due to the underlying prescribing pattern.
Insulin
There are no randomized trials of insulin therapy with fracture or BMD outcomes. Most observational studies have identified increased fracture risk in those using insulin [29–31, 36–38] although others have not found an increased risk [28, 33]. Insulin treatment is also associated with a higher risk of falls [39, 40], and this is likely a contributing factor to the increased fracture risk. Insulin does not appear to have a negative effect on bone; indeed, preclinical studies suggest an anabolic effect. Increased falls and fractures may be the result of more frequent episodes of hypoglycemia and greater frailty due to diabetic complications.
Thiazolidinediones (Rosiglitazone, Pioglitazone)
TZDs increase insulin sensitivity via activation of peroxisome proliferator-activated receptor (PPARγ). Two TZDs, rosiglitazone and pioglitazone, have been used clinically since 1999. A number of studies showed superior efficacy of TZDs over other available antidiabetic therapies in the control of diabetic hyperglycemia [41]. However, their prolonged use is associated with several adverse effects. Strong clinical evidence points to the connection between rosiglitazone use and a significant increase in risk of myocardial infarction and death from cardiovascular causes [42]. This association resulted in a recent review of rosiglitazone safety by the FDA and recommendation for its restricted use in the United States. Interestingly, pioglitazone use is associated with a significantly lower risk of death and lower number of myocardial infarction and stroke incidence [43], indicating that cardiovascular effects of TZDs are not a drug class effect, but rather specifically associated with the TZD type. However, increased risk of bladder cancer in long time pioglitazone users resulted in recent restriction of its use by FDA. Both TZDs exhibit drug class properties of fluid retention and weight gain [44]. Although the use of both rosiglitazone and pioglitazone is currently restricted, the new TZDs with better safety profile are in development. Therefore, understanding TZDs mechanism of action on bone is needed in respect to improvement of safety for bone of new line of TZDs.
Although they possess beneficial anti-hyperglycemic profiles, rosiglitazone and pioglitazone use is associated with adverse effects on the skeleton [45, 46]. The crucial clinical evidence of a causal connection between TZD therapy and increased fracture risk was determined from secondary analyses of results from randomized clinical trials of rosiglitazone and pioglitazone. The first demonstration of increased fracture risk was reported from ADOPT (A Diabetes Outcome Progression Trial), designed to compare time to monotherapy failure of rosiglitazone, metformin and glyburide in recently diagnosed T2DM patients [41]. Because of growing concern, based on rodent models and clinical trials, that TZDs might have a negative effect on bone, the investigators undertook a post hoc analysis of fracture rates in the three groups, using adverse event reports to identify fractures. In 1840 women and 2511 men with a median follow-up of 4.0 years and an average age of 56 (SD 10) years, fracture rates in men did not differ across treatment groups [26, 41]. However, in women, the cumulative incidence of fractures at 5 years was 15.1 % (11.2–19.1) with rosiglitazone, 7.3 % (95 % CI 4.4–10.1) with metformin, and 7.7 % (95 % CI 3.7–11.7) with glyburide, representing hazard ratios of 1.81 (95 % CI 1.17–2.80) and 2.13 (95 % CI 1.30–3.51) for rosiglitazone compared with metformin and glyburide, respectively. Increased fracture rates were seen in the lower and upper limbs. The incidence of hip and clinical vertebral fractures did not differ across treatment assignments, but only four hip and three clinical vertebral fractures were reported in women, as expected in the age range of this trial. Rosiglitazone was associated with higher fracture rates in both pre-and postmenopausal women, suggesting that estrogen status does not modify the effect on bone.
Soon after the ADOPT findings were published, Takeda performed a meta-analysis of pioglitazone trials and reported a similar pattern of increased fracture risk in women, but not men [47]. These observations were subsequently corroborated by other randomized trials. An early meta-analysis of data from ten different randomized controlled trials confirmed that TZD use doubles the risk of fractures exclusively in women [48]. More recently, a meta-analysis of 22 randomized controlled trials, including 896 fracture events, reported increased fracture incidence in women (OR = 1.94; 95 % CI 1.60–2.35) but not in men (OR = 1.02; 95 % CI 0.83–1.27) [49]. Effects in women were similar for rosiglitazone (OR = 2.10; 95 % CI 1.61–2.51) and pioglitazone (OR = 1.73; 95 % CI 1.18–2.55).
Because the TZD trials have included few hip or vertebral fractures, it is necessary to rely on observational studies to assess whether TZD use increases fractures at these particular skeletal sites. A study using registry data in Scotland focused exclusively on hip fracture risk and reported increased risk with greater cumulative TZD use among those with any use (OR per year of exposure 1.18; 95 % CI 1.09–1.28) [33]. Results were similar for pioglitazone and rosiglitazone considered separately. In contrast to reports from randomized trials, increased hip fracture risk was found in men (OR per year of exposure 1.20; 95 % CI 1.03, 1.41) as well as women (OR per year of exposure 1.18; 95 % CI 1.07, 1.29). Those with >4 years of TZD use had OR for hip fracture of 1.94 (95 % CI 1.28, 2.94), compared with those who used a TZD for up to 2 years. Finally, results were similar when evaluated in a subset with adjustment for use of other antidiabetic medications (insulin, metformin, or sulfonylurea). A large observational study using the UK General Practice Research Database also concluded that TZD use increased hip fracture incidence (Rate Ratio 2.09; 95 % CI 1.29–3.40) as well as spine fracture incidence (RR 2.72; 95 % CI 1.29–5.73) [50]. This study reported similar increases in risk of any fracture in men (RR 1.44; 95 % CI 1.18–1.77) and women (RR 1.42; 95 % CI 1.20–1.69).
The principal mechanism underlying increased fracture risk with TZD use appears to be bone loss. A recent meta-analysis of ten randomized clinical trials that assessed change in BMD reported greater bone loss at the lumbar spine, total hip and femoral neck in women randomized to TZD treatment compared with placebo or other antidiabetic medication [49]. Only one trial included men.
Clinical studies of changes in bone turnover markers with TZD treatment have not provided consistent results. In the largest study to date, 12-month changes in serum markers were assessed in 1605 participants in ADOPT [27]. This analysis showed modest but statistically significant increases in levels of resorption marker C-terminal telopeptide (CTX) in women on rosiglitazone therapy compared with glyburide (10.7 % difference, p = 0.002) or metformin (7.3 % difference, p = 0.029). In men, CTX was elevated in the rosiglitazone group compared with metformin (12.2%, p < 0.001) but not compared with glyburide. Both genders had modest reductions in levels of the marker of bone formation P1NP (women −4.4 %, men −14.4 %), but those in the metformin arm experienced greater reductions. For women, changes in P1NP did not differ between the rosiglitazone and glyburide groups while in men losses were greater in the rosiglitazone group. Although rodent models suggest an important role for reduced bone formation as a mechanism of bone loss with TZD treatment, the ADOPT results instead indicate that increases in bone resorption may explain at least in part the increased fracture rate in women on TZD therapy [27].
Smaller trials of rosiglitazone treatment have also reported relative increases in markers of bone resorption compared to placebo [51, 52] and to metformin [53]. However, others have reported a relative reduction in bone formation markers with rosiglitazone treatment, compared with placebo [54] or with diet only treatment [55], and others have reported no difference [56]. For pioglitazone, the largest trial included 156 postmenopausal women with prediabetes and found no differences in bone turnover markers after 12 months compared with placebo [57]. In contrast, a trial in 71 diabetic men reported relative increases in markers of bone resorption (CTX) and formation (P1NP) in the pioglitazone group compared with metformin [58] while a trial in 86 diabetic men and women found a relative increase in a formation but not a resorption marker with pioglitazone treatment compared with placebo [59].
Taken together, results of available clinical studies indicate the following regarding TZD use (1) women are at increased risk of fractures; however, some studies point to elevated risk in men as well; (2) the increased fracture risk appears to be a class effect of currently available TZDs; (3) bone loss is an underlying mechanism; (4) fracture risk is increased in the extremities and most likely at the hip and spine as well.
Mechanism of TZD-Induced Bone Loss
PPARγ, an essential regulator of lipid, glucose, and insulin metabolism [10], is a target for TZDs. The PPARγ protein is expressed in mice and humans in two isoforms, PPARγ1 and PPARγ2. PPARγ1 is expressed in a variety of cell types, including cells of hematopoietic lineage macrophages and osteoclasts [60], whereas PPARγ2 expression is restricted to cells of mesenchymal lineage adipocytes [61]. In bone, PPARγ2 plays an important role in regulation of MSC differentiation toward osteoblasts and adipocytes, and the maintenance of bone mass. Activation of the PPARγ2 isoform with rosiglitazone converts cells of osteoblast lineage to terminally differentiated adipocytes and irreversibly suppresses both the osteoblast phenotype and osteoblast-specific gene expression. Thus, in MSCs PPARγ2 acts as a positive regulator of adipocyte differentiation and a dominant-negative regulator of osteoblast differentiation [11, 62]. In contrast, PPARγ1 expressed in HSC promotes osteoclast differentiation and bone resorption [60]. It controls an expression of c-fos protein, an important determinant of osteoclast lineage commitment and development.
An essential role of PPARγ in maintenance of bone homeostasis was demonstrated in several animal models of either bone accrual or bone loss depending on the status of PPARγ activity [63–68]. In models of bone accrual, a decrease in PPARγ activity in either heterozygous PPARγ-deficient mice or mice carrying a hypomorphic mutation in the PPARγ gene locus led to increased bone mass due to increased quantity of osteoblasts [66, 68]. Interestingly, mice deficient in PPARγ expression in cells of hematopoietic lineage develop osteopetrosis and are less sensitive to the TZD-induced bone loss than control mice [60]. In contrast, in rodent models of bone loss due to PPARγ activation, administration of rosiglitazone resulted in significant decreases in BMD, bone volume, and changes in bone microarchitecture [63, 67, 69]. Observed bone loss was associated with expected changes in the structure and function of bone marrow, which included decreased number of osteoblasts, increased number of adipocytes, and increased support for osteoclastogenesis. The degree of bone loss in response to rosiglitazone correlated with the animal age and the level of PPARγ expression. In younger animals with less PPARγ, bone loss was less extensive than in older animals [69]. Moreover, age determined the mechanism by which bone loss occurred. In younger animals it occurred due to decreased bone formation, whereas in older animals due to increased bone resorption [69]. In addition, studies of rosiglitazone effects in estrogen deficient rats showed that bone loss occurred mainly due to increased bone resorption [64]. In conclusion, animal studies suggest that aging and estrogen deficiency confound TZD-induced bone loss and determine its mechanism.
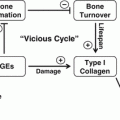
The negative effect of TZDs on osteoblastogenesis includes decreased activity of Runx2, Dlx5, and Osterix, which are osteoblast-specific transcription factors, and decreased activity of osteoblast-specific signaling pathways controlling bone homeostasis, among them Wnt, TGF-β/BMP, and IGF-1 [70, 71]. The effect of TZDs on the expression of genes essential for osteoblast development is strikingly similar to changes observed during aging. Due to the type of bone loss and similarities to aging, some speculate that TZDs may accelerate the aging of bone [69, 72]. The complexity of TZDs effects on bone remodeling resulting from changes on osteoblast and osteoclast differentiation and alterations in bone marrow milieu supporting remodeling are summarized in Fig. 7.3.
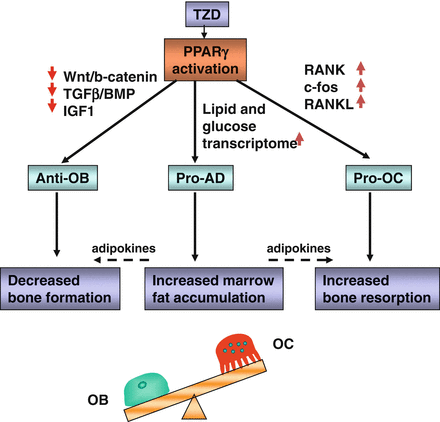
Fig. 7.3
PPARγ activation with TZDs leads to multiple direct and indirect effects in the bone marrow which result in changes in bone cell differentiation, unbalanced bone remodeling and ultimately bone loss. Anti-OB PPARγ activity inhibiting osteoblast differentiation, pro-AD PPARγ activity stimulating adipocyte differentiation, and pro-OC PPARγ activity stimulating osteoclast differentiation
Novel Selective PPARγ Modulators with Beneficial Effect of Insulin Sensitizers and No Effect on Adipocyte Differentiation
The PPARγ ligand-binding domain contains a large binding pocket capable of encompassing a variety of ligands. This provides a wide array of potential contact points that can result in various PPARγ post-translational modifications (PTMs), including phosphorylation, acetylation and sumoylation, and differential recruitment of coactivators, which determine specific activities of this nuclear receptor [73]. The molecular studies provide evidence for distinct mechanisms regulating the proadipocytic, antiosteoblastic, and insulin sensitizing activities of PPARγ and include the levels of Serine 273 and Serine 112 phosphorylation and functional interaction with other proteins such as β-catenin and molecular chaperons FKBP51 and PP5 [74–77].
The concern of TZDs adverse effects has prompted pharmaceutical efforts to develop selective PPARγ modulators which will retain high potency to treat diabetic disease with minimal adverse effects [78]. The PPARγ selective activators, with a decreased proadipocytic activity but intact insulin sensitizing activity such as netoglitazone, INT131, MSDC-0602 and telmisartan do not affect bone mass in mice treated with the therapeutic doses [79–82]. A new class of insulin sensitizers with structural similarities to telmisartan, which block Serine 273 phosphorylation but do not stimulate PPARγ transcriptional proadipocytic activity, has been recently developed [74, 83], however their safety for bone is not as yet determined.
Sulfonylureas
Sulfonylureas function as insulin secretagogues. This class of drugs activates sulfonylurea receptors on the surface of pancreatic β cells and stimulates exocytosis of insulin from vesicles. In addition, sulfonylureas are associated with greater frequency of hypoglycemia which may increase the risk of falls and fractures [84].
In the ADOPT trial, described earlier, fracture incidence was similar in those randomized to a sulfonylurea (glyburide) versus metformin [41]. The results of observational studies have been inconsistent with reports of increased [34], decreased [28, 31] and no difference [29, 30, 32, 35, 50, 85, 86] in fracture risk among those using a sulfonylurea. As noted in a recent review of current literature [87], although a large number of studies have reported no association with fracture, the majority of these studies were not specifically intended to assess the impact of sulfonylureas in particular on fracture. Allocation bias is an important consideration in observational studies, and results from a study in Scotland suggest that patients using sulfonylureas tend to have a higher background fracture risk [33]. Although sulfonylureas increase hypoglycemic episodes, an observational study conducted among Kaiser Permanente members did not find an association between sulfonylurea use and incident falls, identified through inpatient and outpatient medical records [88].
Incretin Analogs and DPP4 Protease Inhibitors
This newest class of antidiabetic drugs enhances the mechanism by which enteric hormones stimulate insulin release from β-cells and inhibit glucagon production in the liver [89]. Glucose-dependent insulinotropic peptide (GIP), and glucagon-like peptides (GLP-1 and GLP-2), are released by gut endocrine cells in response to nutrient intake. Bioactivity of incretin hormones is limited by their rapid degradation and inactivation by dipeptidyl peptidase-4 (DPP-4), a serine protease that is present in a soluble form in plasma and is expressed in most tissues [90]. Recently, incretin mimetics (GLP1 receptor agonists) and DPP-4 inhibitors have emerged as a new class of pharmacological agents to enhance incretins action and improve glycemic control in patients with T2DM. Incretin mimetics and DPP-4 inhibitors have a major advantage over other diabetic medications in that glucose control remains stable with little or no rise in HbA1c levels after long periods of use. The side effects common for incretin-based therapies, including incretin receptors agonists and DPP-4 inhibitors, consist of gastrointestinal, immune system and pancreatic reactions. Since DPP-4 enzyme is known to be involved in the suppression of certain malignancies, particularly in limiting the tissue invasion of tumors, there is a concern that DPP-4 inhibitors may allow some cancers to progress, however clinical data are not as yet available [91–93].
Nutritional hormones are known to be important in bone turnover; as soon as a meal is ingested, bone breakdown is suppressed [94, 95]. Osteoblasts and osteoclasts express receptors for both GIP and GLP incretins. A number of studies indicate that GLP-2 acts mainly as an antiresorptive hormone [96], while GIP can act both as an antiresorptive and anabolic hormone [97, 98]. Mice deficient in GLP-1 receptor develop cortical osteopenia and have more fragile bone as well as increased quantity of osteoclasts and increased bone resorption [99]. GLP-1 receptor signaling may play an essential role in the control of bone resorption indirectly, through a calcitonin-dependent pathway. Calcitonin treatment effectively suppressed bone resorption markers in Glp-1r(−/−) mice, and the GLP-1 receptor agonist exendin-4 increased calcitonin gene expression in the thyroid of wild-type mice [99]. Interestingly, although animal studies showed that DPP-4 inhibitor sitagliptin did not affect bone density, however the absence of DPP-4 in Dpp-4(−/−) mice lead to the greater bone loss after ovariectomy as compared to animals with unaltered DPP-4 expression [100]. In summary, a number of animal studies indicate that incretins have beneficial effects on bone mass and protective effects on bone quality. Therefore, antidiabetic therapies which increase GIP and GLP hormone levels and their bioactivity might exert beneficial effects on human bone.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


