FIGURE 37-1. Obesity and adipocyte hypertrophy. Adipocyte hypertrophy is a key feature of the insulin-resistant state. The flux of free fatty acids out of adipose tissue is disordered in obesity, especially when adipocyte hypertrophy is present. In addition, hypoxia, inflammation, and endoplasmic reticulum (ER) stress have been implicated as important mechanistic links between obesity and insulin resistance. This figure attempts to reconcile the available data on how obesity might lead to insulin resistance. In many instances, the directionality of the connections between mechanistic factors is unclear. For example, it is clear that ER stress can lead to the secretion of inflammatory cytokines, but it is not entirely clear whether inflammation and macrophages might lead to ER stress. Similarly, reduced capillary density (rarefaction) leads to hypoxia, and hypoxia leads to the secretion of chemokines and inflammatory cytokines and probably disordered adipokine secretion. Hypertrophic adipocytes secrete the inflammatory peptide SAA, which may serve to amplify inflammation and chemotaxis of macrophages.342,343 At this time, it is difficult to disentangle the web of deleterious connections. It appears that once the cascade is initiated, multiple interlocking pathways may sustain the dysfunctional adipose tissue.
FIGURE 37-2. Angiogenesis and innervations in adipose tissue. Adipose tissue has the capacity to grow new blood vessels.
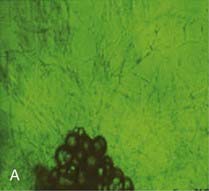
A, Adipose tissue fragments show outgrowth of new capillaries after 10 days culture ex vivo.
(Photomicrograph courtesy Dr. Frank Greenway, Pennington Biomedical Research Center; methods as in reference 174.)
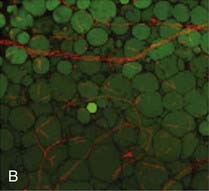
B, In human adipose tissue, capillaries form a rich meshwork around adipocytes.
(Photomicrograph courtesy Drs. Magdalena Pasarica, David Burk, and Steven Smith, Pennington Biomedical Research Center.)


Adipocytes are stained green (BODIPY) and capillaries are stained red (Lectin UEA). In obesity, capillary density is decreased in subcutaneous adipose tissue. Obese (C) adipose tissue and lean (D) adipose tissue have different capillary density. Adipocyte plasmalemma is stained green with Lectin-GS, and capillaries are stained red (Lectin UEA). (Methods are presented in reference 59.)
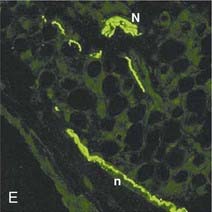
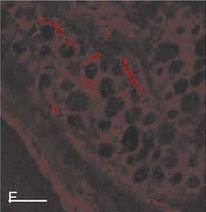
Human adipose tissue is innervated with sympathetic nerve system (SNS) nerve terminals, which are important for the activation of lipolysis. E, Fluorescent immunohistochemistry of nerve fibers (neural specific antibody) in white adipose tissue. F, SNS-specific staining with tyrosine hydroxylase.
(E and F are reprinted from Giordano209 with permission.)
Obesity usually is characterized by increased circulating plasma free fatty acid concentrations. Studies in animals and humans support a causal effect of elevated free fatty acids (produced by lipolysis of triglycerides from adipose tissue) on impaired insulin-mediated glucose metabolism.9–12 Such studies reinforce the original hypothesis proposed by Randle et al in 196313,14 that altered fatty acid metabolism is the key contributing factor to insulin resistance in obese and diabetic patients. Randle and colleagues demonstrated that free fatty acids compete with glucose for substrate oxidation in isolated preparations of heart and diaphragm muscle from rats. More specifically, they proposed that increased fatty acid availability in obesity causes an increase in the intramitochondrial acetyl–coenzyme A (CoA)/CoA and NADH/NAD+ ratios, leading to inactivation of the enzyme pyruvate dehydrogenase. This in turn causes an intracellular increase in citrate concentration, leading to inhibition of phosphofructokinase, the rate-limiting enzyme for glycolysis. As a consequence, glucose-6-phosphate accumulates in the cell and inhibits hexokinase II activity, leading to an increase in intracellular glucose and a decrease in glucose uptake, oxidation, and storage.
Randle’s glucose–fatty acid cycle has been challenged by data from sophisticated clinical studies in which stable isotope turnover was used in conjunction with substrate leg balance methods or 13C and 31P magnetic resonance spectroscopy. These studies provide evidence that insulin resistance in obese people may be related to a primary defect in fatty acid oxidation in skeletal muscle rather than to a competition between fat and carbohydrate metabolism.
First, Wolfe15 identified some of the flaws in the notion that fatty acid availability controls substrate oxidation in the fasting state. He provided evidence that fatty acid oxidation is largely controlled at the site of oxidation, which, in turn, is determined by the availability of glucose. In this model, the primary physiologic role of increased adipose lipolysis in fasting conditions is to provide the necessary glycerol as a gluconeogenic precursor16; the rate of fatty acid oxidation then is regulated by the rate of intracellular metabolism of glucose.
Second, it now is well accepted that skeletal muscle predominantly relies on lipid oxidation during fasting and can easily switch from lipid to increased glucose uptake and oxidation in response to feeding and hyperinsulinemia.17 This switch from fat to carbohydrate oxidation in skeletal muscle has been called metabolic flexibility. It is important to note that Kelley and Mandarino provided convincing evidence that glucose oxidation is increased in the leg of subjects with type 2 diabetes, thereby decreasing its reliance on fat oxidation.18 In opposition to Randle’s glucose–fatty acid cycle, their series of studies suggest that hyperglycemia itself causes an impairment of the normal fasting reliance of skeletal muscle on fatty acids, thereby causing an accumulation of lipids into the muscle tissue. This reversed Randle cycle theory highlights the primary role of impaired lipid oxidation in skeletal muscle rather than excessive lipolysis in the adipose tissue. Growing evidence suggests that the primary cause of the metabolic inflexibility noted in subjects susceptible to insulin resistance is impaired fat oxidation in the fasting state rather than a lack of increased carbohydrate oxidation in response to feeding.19 In support of this concept, evidence now points toward decreased mitochondrial oxidative capacity in insulin-resistant subjects with diabetes20 or a family history of diabetes21 and in insulin-resistant older individuals.22
To prove or disprove the “Randle cycle” hypothesis, Shulman and colleagues23 went one step further. If the Randle hypothesis was true, one would predict an accumulation of glucose-6-phosphate in the skeletal muscle of healthy subjects during glucose and insulin infusions in the presence of high plasma free fatty acid concentration. They directly tested this hypothesis using magnetic resonance spectroscopy. As was expected, high concentrations of circulating free fatty acids caused a reduction in insulin-mediated glucose uptake, with an approximately 50% decrease in glucose storage and a 50% decrease in glucose oxidation.11 However, in contrast to what would be predicted by Randle’s hypothesis, no accumulation of glucose-6-phosphate occurred. Therefore, reduced glucose uptake when free fatty acids are high is due to impaired glucose transport or impaired intracellular signaling. This series of studies shows that intramuscular fatty acids or fatty acid metabolites seem to interfere with the transport of glucose into skeletal muscle cells. Studies have provided some potential mechanisms for the effects of free fatty acid–induced insulin resistance via an impact on insulin signaling at the level of protein kinase C.24–27 More recently, however, Koves et al28 using targeted metabolomics found that obesity-related insulin resistance in skeletal muscle is characterized by excessive β oxidation, impaired switching to carbohydrate during the fasted-to-fed transition, and simultaneous depletion of organic intermediates of the tricarboxylic acid cycle. Therefore, excessive rather than reduced β oxidation underlies the development of muscle insulin resistance by accumulation of acyl-CoAs and their respective acyl-carnitines.28
Although many studies have provided evidence for an association between insulin sensitivity and visceral fat mass (reviewed in reference 29), other studies provide as good evidence for an association between the amount of subcutaneous fat on the trunk and insulin resistance in obese nondiabetic men30,31 and in men with type 2 diabetes.18,32,33 Similarly, insulin resistance in obese women is better related to overall elevated fat mass than to just visceral fat mass.34,35 Thus, subcutaneous fat, which does not drain into the portal vein, causes insulin resistance through a nonportal mechanism. Growing experimental evidence, which does not support the Randle/portal hypothesis, therefore calls for a change in the scientific paradigm to explain the insulin resistance so common in obesity. The bulk of the literature now provides evidence that excessive total fat mass (rather than just visceral fat mass) and impaired muscle fat oxidation are associated with insulin resistance and increased risk for the development of type 2 diabetes. Some of our own studies have clearly shown that gluteal adipose tissue may be protective against insulin resistance rather than precipitating it.36
LINKS BETWEEN OBESITY, INFLAMMATION, AND ER STRESS (see Figs. 37-1 and 37-2)
Studies of murine adipocyte differentiation have revealed an intermediate developmental phenotype between the preadipocyte and the adipocyte that Cousin et al.37 termed the adipiphage, a cell intermediate between the adipocyte and a macrophage. This relationship is not surprising in light of the similarities in gene expression profiles between the two cell types. The macrophage is responsible for consuming extracellular bacteria, cellular debris, and lipids, whereas the adipocyte is responsible for internalizing and sequestering excess lipids. Studies in mice and humans have suggested that adipose tissue contains not only adipocytes and supporting cells, but also macrophages.38,39 The macrophages appear to serve as the major site of TNF secretion and may secrete other cytokines such as interleukin (IL)-8,40,41 an atherogenic cytokine produced in adipose tissue. These studies have also suggested that the bone marrow was the major site of origin for adipose tissue macrophages, indicating that obesity and diabetes might recruit these cells through the production of one or more chemokines.42 The significance of the inflammatory cells is that they, like TNF-α, are likely to activate the NF-κb signaling cascade. Iκκ-β, the upstream activator of NF-κb signaling, plays a key role in insulin signaling and is necessary for the full expression of the insulin-resistant phenotype in the obese ob/ob mouse.43 Upstream activators of this pathway include not only TNF-α, but also fatty acids44 and bacterial lipopolysaccharide. Fatty acids activate this pathway via the Toll-receptor 4, which also responds to lipopolysaccharide.44 In vivo studies in mice43 and humans45 demonstrate that salicylates, inhibitors of the Iκκ-β pathway,46 play an important therapeutic role in insulin resistance and diabetes,47 in part through adipocyte-mediated pathways.48,49 The antidiabetic peroxisome-proliferator–activated receptor (PPAR-γ) ligands (e.g., thiazolidinediones [TZDs]) also decrease the gene transcriptional effects of the Iκκ-β pathway.50
Many more inflammatory cytokines such as monocyte chemoattractant protein-1 (MCP-1) and plasminogen activator protein (PAI) have been linked to adiposity. The increase in inflammatory cytokines found in obesity ultimately characterizes excess adipose tissue as a state of low-grade systemic inflammation, which may link obesity to its comorbidities.51 Obesity therefore occurs in association with an increase in inflammatory cytokines and an infiltration of macrophages within the adipose tissue. Two of the major unsolved questions regarding this observation are these: Why does inflammation occur in parallel with obesity? What is the consequence of this inflammation? One of the growing hypotheses first proposed by Trayhurn’s group52 and recently reviewed53,54 is that inflammation is likely the result of reduced oxygenation (hypoxia; see Figs. 37-1, 37-2, and 37-3) in adipose tissue, which may provide cellular mechanisms for macrophage infiltration, reduced adiponectin secretion, increased leptin secretion, adipocyte death, ER stress, and mitochondrial dysfunction55 in white adipose tissue in obesity.56–58 Inhibition of adipogenesis and triglyceride synthesis by hypoxia may be a mechanism for elevating plasma free fatty acids and reducing adiponectin concentrations, both of which lead to insulin resistance in obesity.56,57 The biological basis for adipose tissue hypoxia could be related to a reduction in arterial blood flow as a result of adipocytes outgrowing their blood supply (Fig. 37-3). A failure to maintain blood flow or a reduction in capillary density (lack of compensatory angiogenesis) or vasoconstriction may serve as the basis for the reduction in blood flow observed in human adipose tissue.53 The hypothesis that increased adipose tissue mass in obesity without adequate support of vascularization might lead to hypoxia, macrophage infiltration, and inflammation was recently confirmed in human studies.59
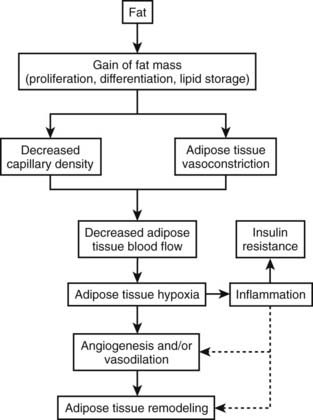
FIGURE 37-3. Hypoxia, inflammation, and insulin resistance.
(This figure is adapted from a recent review by Ye.53)
Rapid growth of adipose tissue leads to an expansion that may not be accompanied by a simultaneous maintenance of capillary density through increased angiogenesis or vasodilation and therefore a decrease in blood flow to the tissue. Decreased blood flow per unit of adipose tissue leads to tissue hypoxia, which, in turn, induces inflammation. When inflammation becomes out of control with increased systemic and local cytokine concentrations, insulin resistance occurs. Adipose hypoxia therefore is a signal for the remodeling of adipose tissue.
Over the past few years, publications have provided growing evidence suggesting that ER stress may play a role in the pathogenesis of type 2 diabetes.60 The ER is a highly dynamic organelle with a central role in lipid and protein biosynthesis. It produces the transmembrane proteins and lipids for most cell organelles and is responsible for the synthesis of almost all secreted proteins. After translation of proteins is performed by ribosomes on the cytosolic surface of the ER, the unfolded polypeptide chains are translocated into the ER lumen, where numerous chaperone proteins are crucial for the proper folding of proteins and protein complexes.61
However, the ER is exquisitely sensitive to alterations in homeostasis, and proteins formed in the ER may fail to attain proper conformation owing to (1) lack of chaperones or energy to promote chaperone–protein interactions, (2) calcium depletion, (3) disruption of the redox state, (4) protein mutations that hamper adequate folding, and (5) reduction of disulfide bonds.60 ER stress is also called the unfolded protein response (UPR). Lipotoxicity and glucotoxicity as seen in prediabetes and diabetes are triggers of ER stress.
Excess free fatty acids as seen in obesity activate the ER stress response in β cells, eventually causing apoptosis.62 Recent data suggest that hypoxia in adipose tissue of obese mice and people may contribute to the induction of ER stress, thereby affecting adipokine production. For example, adiponectin expression was decreased in adipose tissue from high-fat–fed KKAy mice.63 Evidence for the role of ER stress in human tissues such as pancreas, muscle, and adipose is presently not available. Similarly, increases in expression and phosphorylation of stress-activated kinases such as p38 and JNK were detected in omental adipose tissue from obese women compared with lean controls.64 Over the next few years, research in this area should lead to a better understanding of the interaction between obesity, inflammation, hypoxia, and ER stress.
LINK BETWEEN TOO LITTLE FAT AND INSULIN RESISTANCE
At the other end of the spectrum from obesity, it is now recognized that a lack of adipose tissue is associated with insulin resistance and increased risk for development of type 2 diabetes (see Chapter 38). Lipodystrophy in humans is an acquired or hereditary syndrome characterized by decreased adipose tissue mass, insulin resistance, and often diabetes mellitus.65–68 In these patients, insufficient adipose tissue mass leads to excess energy storage as triglycerides in liver and skeletal muscle and causes insulin resistance in these tissues.69,70 Genetic manipulation causing ablation of adipose tissue in mice supports the link between adipose deficiency and insulin resistance. Transgenic animals without adipose tissue store lipid in skeletal muscle and liver and develop insulin resistance, glucose intolerance, and eventually diabetes.71–73 This is identical to the fatty liver and muscle seen in obesity and type 2 diabetes. Furthermore, transplantation of adipose tissue back into lipoatrophic animals reverses the elevated glucose levels.74 However, transplantation of adipose tissue from leptin-deficient mice (ob/ob) did not improve the metabolic abnormalities, indicating that the sequestration of triglyceride into adipose tissue is not entirely sufficient to restore insulin sensitivity.75 In humans, treatment of lipodystrophic patients with leptin can dramatically reverse the fatty liver and insulin resistance.76–78 On the other hand, surgical removal of adipose tissue causes the metabolic syndrome.79 Together, these studies demonstrate that, as in obesity, inadequate adipose tissue mass leads to ectopic fat storage and metabolic disturbances. Too little fat therefore is as deleterious as too much fat and predisposes to the development of the metabolic syndrome with insulin resistance and ultimately type 2 diabetes. Three new paradigms may explain insulin resistance:
Obesity Is Another Ectopic Fat Storage Syndrome
Positive energy balance in our “obesigenic” environment produces a pattern similar to lipodystrophy in humans, that is, excess lipid storage in liver80 and skeletal muscle,23,81,82 followed by insulin resistance, glucose intolerance, and diabetes. However, in contrast to lipodystrophic patients, adipose tissue stores are adequate or even large in obese patients, suggesting that the size of adipose tissue becomes inadequate to sequester dietary lipid away from liver, skeletal muscle, and pancreas. The adipocyte becomes hypertrophic and is unable to recruit and/or differentiate new adipocytes to store the excessive dietary fat.83 The Danforth hypothesis is supported by the fact that, independent of total fat mass, individuals with larger fat cells are at higher risk of developing type 2 diabetes than are individuals with smaller fat cells.84 Furthermore, TZDs improve insulin resistance in part by promoting the differentiation of new fat cells in subcutaneous adipose tissue through activation of PPAR-, thereby providing extra storage capacity for dietary fat.85,86 Adipogenesis translates into a gain in subcutaneous adipose tissue87 and a decrease in lipid infiltration in skeletal muscle and liver.88,89 Through the upregulation of genes in the lipid storage and synthesis pathways in adipose tissue, TZDs also decrease free fatty acids, providing a second mechanism for protection of liver, muscle, and the cell from fatty acids.90–92 As is discussed later in this chapter, drugs may be designed to decrease ectopic fat storage by increasing adipogenesis and/or increasing fat oxidation, leading to improved insulin action. Weight gain in humans is probably due to an increase in food intake consistent with the action of the PPAR-γ agonists in increasing food intake.93,94
Adipose Tissue: Hypertrophy vs. Hyperplasia
Historically, adipose tissue was viewed as an inert tissue with a singular function: lipid storage. The main areas of research in the field of adipose tissue were related to adipocyte size and number, as well as to lipid synthesis, adrenergic regulation of lipolysis, and insulin signaling in isolated adipocytes. In adults, obesity is associated with an increase in both the number and the size of adipocytes95,96 (Fig. 37-4). The increase in fat cell size (hypertrophy) is thought to reflect an imbalance between adipocyte lipid uptake or synthesis and the release of lipid via lipolytic pathways. In addition to increased adipocyte size, obese individuals have an increase in the absolute number of adipocytes (hyperplasia). Early studies demonstrated heterogeneity in fat cell size; some obese patients have adipocytes as large as 1 mL, and others have very small fat cells. This heterogeneity led to the concept of hypertrophic or hyperplastic obesity based on the average size of fat cells (see Fig. 37-4). In contrast to this dichotomous viewpoint, the reality is that obese individuals cannot be grouped into such simple categories. There is a continuous distribution of fat cell size, and most obese patients have both hypertrophy and hyperplasia. Increased fat cell size is correlated positively with fasting insulin and negatively with insulin sensitivity.97 In Pima Indians, increased abdominal adipocyte size is associated with insulin resistance,84,98 a potential inherited trait99 that predicts the onset of type 2 diabetes.84 Together, these data suggest that increased fat cell size is important to whole body metabolism and insulin action.
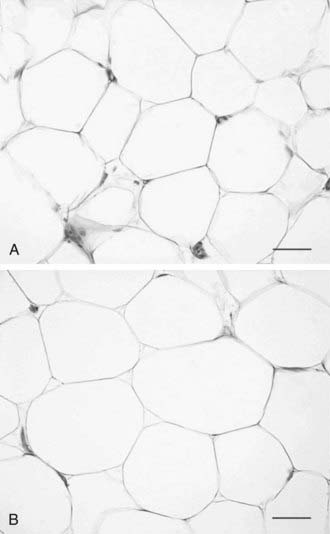
FIGURE 37-4. Hyperplastic versus hypertrophic obesity. Adipocytes are derived from mesenchymal stem cell precursors (see Fig. 37-7). Both mesenchymal precursors and the preadipocyte, whose differentiation potential is limited to the adipocyte lineage, can undergo mitosis. Hyperplastic obesity (A) is defined by an increase in the number of adipocytes, while adipocyte size remains small. Hypertrophic obesity (B) is characterized by larger adipocytes. The reason for the increase in adipocyte size is unclear. Current hypotheses suggest that failure of the large adipocyte to recruit preadipocytes to differentiate may play a role in the development of insulin resistance, a precursor to overt β cell failure as manifested by diabetes.
(Photomicrographs courtesy of Prof. Saverio Cinti, MD, Institute of Anatomy, Faculty of Medicine, University of Ancona, Italy.)
There are several ways of thinking about why fat cells might be large in obese individuals. First, adult adipose tissue has been viewed as a nonmitotic tissue, and increases in adipocyte size might simply reflect an imbalance between storage and lipolysis. If the number of adipocytes is considered fixed, any increase in adipose tissue mass is the result of increased lipid storage in adipocytes. A second view is that fat cells can be recruited continually to differentiate into mature lipid-storing fat cells, and large fat cells are an indication of the failure of this process. Several investigators have proposed that once adipocytes are filled to a certain degree, new fat cells are recruited, and lipid then is stored in these new insulin-sensitive and lipid-hungry adipocytes. The cross-sectional data used to support this model are presented in Fig. 37-5. Average fat cell size increases as body fatness increases up to a certain point, after which increased adiposity does not result in an increase in fat cell size. Even if individuals with hyperplastic or hypertrophic obesity lose weight equally, hyperplastic obese patients regain the weight much more quickly than do hypertrophic subjects, lending support for the concept of small lipid-hungry fat cells100 (Fig. 37-6). During weight loss, fat cell size decreases without a change in fat cell number but with a decrease in fasting insulin.97 This has been interpreted as evidence that once a fat cell is formed, it is permanent. However, recent studies demonstrate a relatively high rate of adipocyte turnover101 and evidence for regulation of apoptosis.102 It should be noted that longitudinal data and precise measures of fat cell number are not sufficient to confirm this model in humans. This in part is the result of our current inability to accurately quantify the numbers of stem cells and preadipocytes in adipose tissue in vitro or in vivo, and it is associated with the difficulties involved in quantifying the very smallest fat cells in adipose tissue.103
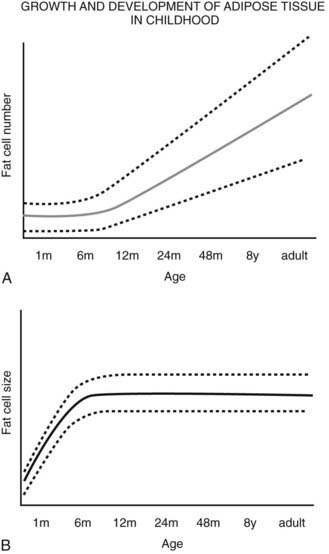
FIGURE 37-5. Schematic relationship between age, body fat mass, and fat cell size/number in childhood. A, Whole body fat cell number remains constant during the first year of life and then increases over time.
A wide range of fat cell size and number across individuals is indicated by the dashed lines. B, The increase in body fat that is seen during the first year of life occurs primarily as a result of increased lipid storage and hypertrophy of existing adipocytes rather than through the recruitment of preadipocytes.
(A and B adapted from the data from Hager105 and Soriguer Escofet.106)
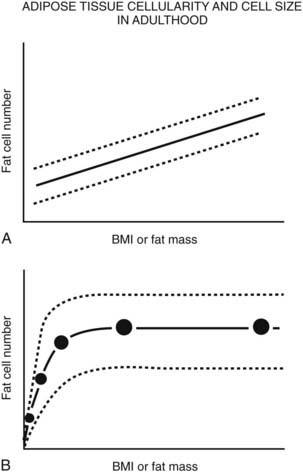
FIGURE 37-6. Schematic relationship between body fat mass and fat cell size/number in adulthood. A, In adults, whole body fat cell number increases with increasing body mass (hyperplasia). B, Again in adults, cross-sectional data show a positive correlation between body fat mass and fat cell size at lower body fat mass (solid line) until fat cell size reaches a plateau at higher levels of fat mass. At this point, fat cell size cannot increase any further because of (1) limitations in lipid storage for unknown reasons, and/or (2) recruitment of existing preadipocytes to differentiate and store lipid, and/or (3) proliferation and differentiation of mesenchymal precursor cells into mature lipid-storing adipocytes. If fat cell proliferation, differentiation, and/or recruitment did not occur, fat cell size would continue to increase as fat mass increased. A wide range of fat cell size and number across individuals is indicated by the dashed lines. Fat cell number does not appear to change with weight gain or loss; however, precise tools are not available to quantify small changes in adipocyte number in vivo in humans.
(Data from Hirsch J, Batchelor B: Adipose tissue cellularity in human obesity, Clin Endocrinol Metab 1976;5:299–311.)
In contrast, the cross-sectional data illustrated in Fig. 37-6 could be interpreted as evidence for the recruitment of new adipocytes. If fat cell hypertrophy was the only way to gain fat, then fat cell size would increase linearly with fat mass. This is not the case. Based on cross-sectional data, the point at which hypertrophy recruits new fat cells probably occurs at a cell volume between 0.8 and 1.0 mL.104–106 Two additional pieces of evidence support the view that recruitment of new fat cells occurs in vivo in humans. First, when adipose tissue is separated into fat cells and the remaining cell populations (stromal-vascular fraction), adipocytic precursor cells from the stromal-vascular fraction are able to differentiate in vitro into mature lipid-storing adipocytes throughout life and into old age. Obesity and age are determinants of the capacity to differentiate adipocytes in vitro.107,108 Recent studies of in vivo DNA synthesis in humans suggest that adipocyte turnover is high in adult humans with a 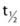 ranging from 240 to 425 days.101,109 In addition, recent work suggests that the large fat cells secrete a factor or factor(s) that promote adipocyte proliferation and differentiation—a finding that is consistent with the recruitment of new adipocytes by hypertrophic adipocytes, as was discussed earlier.110
ranging from 240 to 425 days.101,109 In addition, recent work suggests that the large fat cells secrete a factor or factor(s) that promote adipocyte proliferation and differentiation—a finding that is consistent with the recruitment of new adipocytes by hypertrophic adipocytes, as was discussed earlier.110
Studies by Hirsch and others in rodents demonstrated that animals that were calorically deprived before weaning had a reduced total number of fat cells when compared with animals that were suckled in smaller litters with higher caloric intake. Similarly, about one half of the obesity in Zucker fatty rats can be prevented by early restriction of energy.111 This gave rise to the concept that early overfeeding during adipose tissue development might increase the population of adipocytes and their precursors that produce obesity over time. This model, known as the adipose cell or critical period hypothesis, predicts that a large number of adipocyte precursors early in life could lead to the development of obesity by providing a “sink” destined to be filled with lipid. A corollary to this concept is that individuals with a reduction in adipocyte precursors, similar to those individuals with failure of adipocyte differentiation as described earlier, would be predisposed to the development of diabetes when food intake is increased, as their storage capacity for excess fat is diminished.
Although the concept of an early life critical period for adipocyte precursor development has been much discussed, by comparison the actual data supporting this concept are sparse. Consistent with this concept, obese subjects with an early childhood onset of obesity tend to have smaller fat cells when compared with those with later adult onset of obesity.112,113 Similarly, children of mothers who were energy deprived during the Dutch famine of 1945 had a lower incidence of obesity in adulthood.114 Although no prospective long-term data exist to support the adipose cell hypothesis, the cross-sectional data support the concept that in many cases of early-onset obesity, adipose tissue size tends to be hyperplastic rather than hypertrophic; the latter is seen with late-onset obesity.
The original data and hypothesis presented by Hirsch suggested a single early critical period; later discussions offered the concept that additional critical periods of adipocyte precursor proliferation might exist,96 with recruitment from the precursor pools into mature adipocytes throughout life. At birth, a typical infant has about 4 billion observable fat cells, and this number increases to approximately 10 to 40 billion in lean individuals and up to 50 to 100 billion in obese patients,115 supporting the concept of ongoing adipocyte proliferation and/or recruitment throughout life. In contrast to rodents, humans have a long, slow growth and development period (neotony) and are likely to have several critical periods of adipocyte development.116
Body fat mass increases during the first year of life, most often through fat cell hypertrophy. After the first year, the number of adipocytes increases, the fat cell size remains relatively constant, and whole body adiposity (% fat) decreases. At about 6 years of age, % body fat begins to increase again. This has been termed the adiposity rebound. Longitudinal body weight data in children demonstrate that an earlier adiposity rebound is associated with obesity in adulthood.117–119 Although no detailed information is available on the relative role of hypertrophy versus hyperplasia for this age range, the adiposity rebound is considered a critical period for adiposity later in life.120
Regulation of Adipogenesis
Adipose tissue, unlike other tissues such as brain, kidney, or liver, retains the ability to increase in size in adulthood. Several processes control the mass of adipose tissue in the body:
Adipocytes can be classified based on anatomic location as subcutaneous, visceral (intraperitoneal), bone marrow, and structural (periorbital, palms of the hands and soles of the feet). The hereditary and acquired lipodystrophies teach us that each of these depots of adipose tissue is developed or regulated differently, as each form of lipodystrophy results in loss or failure to differentiate in specific depots. For example, in congenital generalized lipodystrophy, mechanical adipose tissue of the palms and soles is spared.121
Adipose tissue precursors are primarily mesenchymal in origin (Fig. 37-7). These precursor cells, also known as preadipocytes or stromal cells, have the capacity to differentiate into a limited number of cell types, including adipocytes, osteoblasts, and chondrocytes.122 At least two distinct subtypes of preadipocytes have been identified (reviewed in reference 123). These two subtypes, probably distinct from the white adipose tissue/brown adipose tissue switch described later, differ in their capacity for replication, differentiation, and susceptibility to TNF-α-induced apoptosis. Subcutaneous adipose tissue preadipocyte precursors replicate faster and differentiate better than omental precursors. Evidence suggests that omental and mesenteric adipose tissue preadipocytes differ as well.124,125 These differences in preadipocyte characteristics are postulated to influence the propensity of an individual to store fat in the visceral versus subcutaneous depots. Transcriptome analysis of subcutaneous and visceral adipose tissue reveals a distinct pattern of expression of many genes; not only between depots but also across body mass indices (BMI).126,127 In addition, major differences have been noted in the expression of developmental genes in subcutaneous versus visceral adipose tissue. Taken together with the cellular phenotypes identified by Kirkland’s laboratory,128 this suggests that different cell types contribute to differences in the function of each adipose tissue region, and these differences can be explained by developmental programming. In addition to these developments in the different types of preadipocytes, the origins of the preadipocyte are becoming clearer. Recent work involving multiple tissues, including adipose tissue, suggests that pericytes—cells that lie just outside the capillary—are a major source of mesenchymal stem cells and preadipocytes.129,130
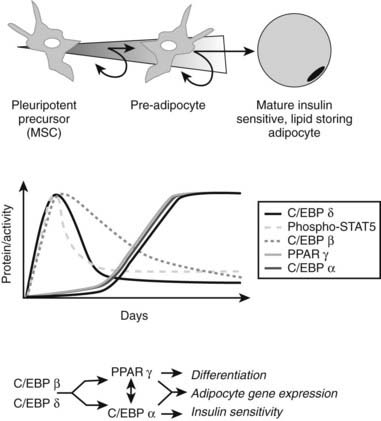
FIGURE 37-7. Adipocyte differentiation cascade. Adipocytes are derived from mesenchymal stem cell precursors (MSCs). Both the mesenchymal precursors and the preadipocyte, whose differentiation potential is limited to the adipocyte lineage, can undergo mitosis. The middle panel depicts the coordinate, sequential activation of the major nuclear transcription factors thought to be involved in the adipogenic differentiation process. PPAR-γ is considered an obligatory “master” regulator of adipogenesis. The PPAR-γ system turns on genes involved in lipid synthesis and insulin action (see text for details).
We know very little about the systems that control proliferation of adipose tissue precursors. Most of what we know is derived from the study of the cellularity of rodent adipose tissue or the behavior of stromal-vascular cultures in vitro. Some of the known activators and inhibitors of adipogenesis are presented in Table 37-1. For example, insulin-like growth factor-1 (IGF-1), a growth factor under the control of insulin and growth hormone in adipose tissue, promotes the proliferation and differentiation of preadipocytes, while transforming growth factor (TGF) inhibits proliferation.131 More is known about the processes whereby adipocyte precursors, particularly 3T3-L1 preadipocytes, proceed along the pathway from precursor to mature adipocyte. We now know that a series of transcription factors coordinately regulate multiple genes in a tightly regulated temporal fashion. As is shown in Fig. 37-7, each of these transcription factors forms a nonredundant network that, once initiated, leads to the emergence of the adipocyte phenotype. Some of the known key transcription factors include PPAR-γ, STAT5, C/EBP/α, and SREBP1c/ADD, CREB, and Wnt/frizzled. In addition, a new family of proteins—the krüppel-like factors—has been identified as regulators of adipocyte differentiation.132
Table 37-1. Extracellular Factors That Regulate Adipogenesis
Multiple hormones, cytokines, growth factors, cell cycle regulators, and adhesion molecules control this differentiation cascade. Classic studies by Green and others showed that when confluent, clonal cell lines such as 3T3-L1 and F442A differentiate into adipocytes if exposed to a cocktail of insulin, dexamethasone, and isomethylbutylxanthine (IBMX).133–135 Emphasis has also been placed on the role of cell cycle and the necessity for proliferation prior to differentiation of precursors. However, recent data suggest that this has more to do with the E2F transcription factors than with the process of mitosis per se.136,137
IBMX and other agents that increase cyclic adenosine monophosphatase (cAMP) act through the transcription factor CREB.138 Several transcription factors are critical for the conversion of cells from a fibroblastic phenotype to an adipocytic phenotype. PPAR-γ has received the most attention, and this is warranted since overexpression of PPAR-γ into fibroblastic cell types is sufficient to confer the adipocytic phenotype.139 There are several putative endogenous ligands for PPAR-γ, including the prostaglandin PGJ2,140,141 long-chain fatty acids, and 13-HODE and 15-HETE, which can be generated from linoleic and arachidonic acids, respectively, by a 12/15-lipoxygenase.142 All of these compounds can activate the PPAR-γ transcription factor that heterodimerizes with the RXR transcription factor to turn on genes in the glucose uptake,143,144 lipid uptake,145 and lipid synthesis pathways.146,147 The true endogenous ligands are unknown, but their synthesis/activity appears to be downstream of the C/EBP-β transcription factor.148 C/EBP-α is expressed contemporaneously with PPAR-γ and facilitates the full adipocytic phenotype. Immediately upstream of PPAR-γ lie the C/EBP transcription factors C/EBP-β and C/EBP-δ, which upregulate PPAR-γ. Other transcriptional promoters of adipogenesis include STAT 5,149 the glucocorticoid receptor, and ADD/SREBP-1c.150 Transcriptional inhibitors include GATA 3,151 TCF/LEF, and the Wnt pathway.152 Combined with the transcriptional activators, they cooperate in an orchestrated cascade of transcriptional events leading to a mature adipocyte.
Last, PPAR-γ cofactors may regulate the ultimate transcriptional program in adipocytes. For example, adipocytes can be converted from energy storage to energy consumers by the PPAR-γ cofactor PGC-1a.153 Similarly, the PPAR-γ cofactors SRC-1 and TIF2 may determine the responses of adipose tissue to high-fat diets, with SRC-1 activating fatty acid oxidation and TIF2 promoting lipid storage.154 These two examples highlight a growing understanding that not only the ligand but also the transcription factors and cofactors are important in whole body metabolism. The intracellular transcriptional control system is regulated by extracellular signals from cytokines, hormones, neural inputs, and the autocrine/paracrine production of ligands for these transcription factors.
Adipocytes and indeed virtually all other cells store neutral lipids such as triglycerides in droplets of varying sizes. These droplets are formed as triglyceride is synthesized in the endoplasmic reticulum or is taken into the cell from the plasma membrane. Lipid droplets are coated by a layer of phospholipids and proteins that serve to sequester the neutral lipids from the cytosol and to regulate the access of lipases to the surface of the lipid droplet. Substantial progress has been made in the identification of these lipid coat proteins since the initial discovery of perilipin and other members of the perilipin family (Perilipin, Adipophilin, S3-12, and TIP-47 [PAT]).155 Consistent with their functional role, the structure of these proteins is highly conserved with both hydrophilic and hydrophobic domains. In adipose tissue, each PAT protein plays a distinct role, contributing to lipid synthesis or lipolysis. For example, perilipin-A is phosphorylated by PKA and PKG, which allows docking of lipases and the initiation of lipolysis.156 On the synthesis side, the repertoire of lipid droplet proteins changes as the lipid droplet is formed and matures, demonstrating the on-off exchange of these proteins and the dynamic nature of the process for formation and movement of lipid droplets. Once again, the biology is showing us that the adipocyte is not a static, inert tissue but rather participates as an active organ in the regulation of metabolism via lipid droplets, which are dynamic organelles in their own right. Lipid droplet proteins may play an important role in lipid oxidation independent of their role in lipolysis. A novel PAT protein, LDRP5 (also called OXPAT or Mldp) appears to be important for activating lipid oxidation in oxidative tissues such as cardiac myocytes.157
Given their central role in lipid storage and synthesis, the PAT proteins are being actively explored as a means of improving our understanding of the regulation of these critical cellular processes.
BROWN ADIPOSE TISSUE IN HUMANS
Brown adipose tissue (BAT) is an exquisitely designed tissue/organ system that has evolved for the maintenance of body temperature. It is characterized by smaller cells with large quantities of mitochondria and small lipid droplets, providing the potential for high cellular metabolism. At the metabolic, protein, and transcription levels, the BAT is upregulated principally by the sympathetic nervous system when production of heat is needed to maintain body temperature. Years ago, Nicholls and Ricquier158,159 described a mechanism for heat production based upon a specific highly abundant protein (uncoupling protein 1; Ucp1) in the inner mitochondria of brown adipocytes that uncouples the production of chemical energy as adenosine triphosphate (ATP) from oxidative phosphorylation and instead produces heat.
Until recently, BAT has been thought to be most important for maintaining body temperature in small mammals and infants, but a function in the physiology of adult humans was dismissed because of low numbers of brown adipocytes.160,161 However, unrelated pursuits in nuclear medicine using positron emission tomography (PET)/computed tomography (CT) scanning techniques have revealed the presence of BAT in adult humans, especially after cold exposure.162,163 The question now is how can we induce this amazing organ not only to generate heat but also to enhance fat oxidation and thereby reduce obesity.
Although the normal function of brown fat thermogenesis may be specific for the regulation of body temperature, many genetic and pharmacologic studies in rodents have shown that constitutive overexpression of Ucp1 in white fat and skeletal muscle can drastically reduce both genetic and diet-induced obesity, offering therefore a new safe molecular target for the treatment of obesity.164–166 This potential for brown fat adaptive thermogenesis as a drug target for obesity has not been ignored by the pharmaceutical industry. Unfortunately, many candidate agonists of the β3-adrenergic receptor have failed in human clinical trials, even though these drugs have been efficacious in rodent models of obesity. What is different in the human and in the mouse?
Most of the effects of genetic, pharmaceutical, or cold-induced upregulation of Ucp1 in the mouse models result in the emergence of new brown adipocytes in white fat depots with levels of Ucp1 upregulated several-hundred fold (Fig. 37-8). Unfortunately, human white adipose tissue does not appear to be able to mount such transient induction of brown adipocytes. The failure of brown fat thermogenesis in humans appears to be based on the lack of fundamental information on the mechanisms controlling the developmental origins of brown adipocytes within discrete brown fat depots (e.g., interscapular brown fat) and on the small number of diffusely localized brown adipocytes in various white fat depots. However, a recent paper from the Spiegelman group167 elegantly describes a novel transcription factor PRDM16, whose presence can promote the differentiation of preadipocytes and myoblasts into brown adipocytes, and whose absence promotes the myogenic differentiation program (see Fig. 37-8). It is important to note that the ability of PRDM16 to induce the brown adipocyte lineage is restricted to discrete brown fat depots, such as those found in the interscapular region, but it does not participate in induction of the diffuse brown adipocytes located in the white fat depots. The data support the concept that interscapular BAT and brown adipocytes in white fat have separate independent developmental origins.168 PRDM16 is clearly an important player in brown adipogenesis but may not be sufficient, since PRDM16 KO mice have significant levels of interscapular fat with Ucp1 expression.167 However, one does not know whether upregulation of PRDM16 in humans can induce increased discrete brown adipose tissue and/or diffuse brown adipocytes.
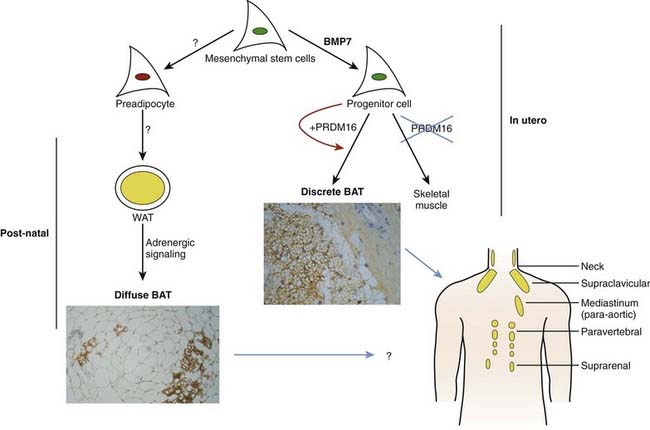
FIGURE 37-8. Brown adipose tissue in humans. Mesenchymal stem cells can have two major pathways, one into preadipocytes and another into progenitor cells of brown adipocytes or skeletal muscle cells. Bone morphorgenetic protein 7170 activates a full program of brown adipogenesis, including induction of early regulators of brown fat PRDM16 and PGC-1α.167 In absence of PRDM16, these progenitor cells are transformed into skeletal muscle cells. Preadipocytes are differentiated into white adipocytes in subcutaneous and visceral adipose tissue. By stimulation of adrenergic signaling, some of these white adipocytes can be transformed into brown adipocytes defused in the white adipose tissue. On the other hand, discrete brown adipocytes are concentrated in brown adipose tissue depots such as in the neck and supraclavicular, paravertebral, and suprarenal regions. The recently discovered brown adipocyte tissue regulator PRDM16167 is responsible, at least in rodents, only for the generation of discrete brown adipose tissue.
Although the regulator PRDM16 has provided important insights into the developmental origins of discrete brown fat depots, the next important step will be to determine the origin(s) of diffusely localized brown adipocytes in white fat depots at least in rodents. Diffuse BAT adipogenesis is more closely related to increased thermogenesis and reduced obesity.127 Enthusiasm for the promise of PRDM16 as a drug target needs to be tempered by the caveat that mice with an inactivated PRDM16 gene die at birth, suggesting that PRDM16 is a transcription factor with additional unknown functions in mammalian development. It has long been known that chronic increases in circulating catecholamines in patients with pheochromocytoma lead to large brown fat depots.169 These historical data, together with recent findings of discrete brown fat depots uncovered by PET technologies,162 should stimulate a renewed effort to find strategies to induce an increase in brown adipocytes, and to ask why the many previous studies with β3-agonists failed to significantly stimulate thermogenesis in humans. Maybe the lack of β3-adrenergic receptors in human white adipocytes is something that needs to be overcome to facilitate conversion of white to brown adipose tissue and to stimulate thermogenesis. It is also important to evaluate the effects of BMP7 on stimulating the enhanced expression of brown adipocytes, as recently shown by Tseng et al.170 The discovery of previously uncovered brown adipose tissue/cells in adult humans and its potential physiologic significance in cold- and dietary-induced thermogenesis should help in revamping our effort to target the molecular development of brown adipogenesis for the treatment of obesity.” bib9002 bib9003″>170a–170c
INTEGRATIVE BIOLOGY OF THE ADIPOSE TISSUE
To maintain energy homeostasis, the brain has two avenues of communication with the periphery: hormones and neurons. The adipose tissue is no exception to this rule, since it is well established that both hormones and neurons control its life cycle and metabolism. An understanding of how adipose tissue operates and is regulated in vivo is a prime example of integrative biology. Imaging studies of the different adipose depots, as well as studies of adipose tissue metabolism and secretory function using arterial-venous differences and microdialysis in conjunction with measures of adipose tissue blood flow, have improved our integrative view of the metabolism and function of adipose tissue.
Appreciation of the importance of the vasculature to the proper functioning of the adipose tissue is increasing. Nutrients such as glucose, fatty acids, and proteins are supplied from the vasculature and are critical to the growth and maintenance of adipose tissue. Recent studies have focused on the supply of oxygen for oxidative metabolism in adipocytes. Current data suggest that the delivery of oxygen may be limiting in adipose tissue of obese mice. The consequences of the hypoxia are unclear, but in vitro data52 support the in vivo data,56 suggesting that hypoxia leads to chemotaxis of macrophages and inflammation. These findings were recapitulated in human adipose tissue.59 Under these circumstances, hypoxia should turn on the transcription factor HIF-1, leading to angiogenesis and a reversal of the hypoxia. This does not appear to be the case in mature adipose tissue,171,172 in which vascular endothelial growth factor (VEGF), a primary downstream transcriptional target of the HIF-1 pO2 sensing system, is not activated. Anatomically, good evidence suggests that the new blood vessels sprout from existing endothelial cells,173 and this has been co-opted in the development of ex vivo assays to clearly demonstrate that human adipose tissue is capable of sprouting/angiogenesis.174 Why the angiogenic signals are not increased when pO2 is decreased is a paradox that needs to be investigated.
In murine adipose tissue development, angiogenesis, adipogenesis, and the stromal cells interact in a way that is coordinated175,176 and relies on VEGF.177 This is different from the situation in mature hypoxic adipose tissue, highlighting the differences in mechanisms and the relative importance of growth factors in growth and development as opposed to the events that occur in mature adipose tissue. Adiponectin, secreted by small but not hypertrophic inflammatory adipocytes, stimulates angiogenesis, highlighting the interplay of adipokines, which we typically consider metabolic hormones working in concert with the growth factors classically implicated in angiogenesis.178
Endocrine Signals
Glucocorticoids
Glucocorticoid treatment of laboratory animals results in the development of obesity. Animal models of obesity invariably have increased levels of corticosterone. Adrenalectomy results in the reversal or prevention of obesity. Activation of the glucocorticoid receptor results in differentiation of preadipocyte precursors133–135 and lipid storage in adipocytes. In humans, overproduction of cortisol (Cushing’s syndrome) results in a phenotype of central (abdominal) obesity, hypertension, and diabetes. Of the many investigations into the role of adrenal glucocorticoids in human obesity, most show normal urinary free cortisol, normal circadian variation in cortisol values, and normal plasma cortisol values, although metabolic clearance rate and production are increased.179 The enzyme 11HSD-1 is present in human adipose tissue and converts inactive cortisone into active cortisol.180 It is important to note that there is a strong positive correlation between adipocyte size and the activity of 11HSD-1 in converting cortisone into cortisol. This fits with the idea that adipocyte hypertrophy generates signals, like cortisol, to recruit new adipocytes.
The most compelling data for an association between human obesity and cortisol come from studies that classify obese women into central and peripheral types of obesity. By stratifying volunteers on this basis, Marin and coworkers181 demonstrated an increase in urinary and serum cortisol as the waist-to-hip ratio (WHR) increased. Serum cortisol responses to stress were greater in women with high WHR, suggesting a role of response to environmental stressors as a potential factor in abdominal obesity.181,182 Other evidence in humans suggests that cortisol values within normal concentrations are sometimes related to fat patterning, possibly via increased sensitivity to exogenous stressors. Genetic factors also may determine the susceptibility of adipose tissue to these exogenous stressors.183
Growth Hormone/IGF-1
Growth hormone (GH) is a potent lipolytic hormone.184 GH receptors activate classic cAMP lipolytic systems in adipose tissue. In addition to stimulating lipolysis, GH increases IGF-1 production in adipose tissue.185 IGF-1 potently activates preadipocyte proliferation and differentiation of precursors into mature lipid-storing adipocytes.131 Deficiency of GH is associated with central obesity, and replacement of GH reduces visceral adiposity.186 Despite early reports of the therapeutic efficacy of GH in men with central obesity,187 other studies do not show this effect and in fact report an increase in body fat after GH withdrawal compatible with the effects of IGF-1 in promoting adipoctye differentiation.171 GH treatment in the absence of clear-cut GH deficiency cannot be recommended, as the side effect profile includes edema, carpal tunnel syndrome, glucose intolerance, and many others. GH-like peptides that increase lipolysis without upregulation of IGF-1 synthesis have been discovered188 and may be beneficial without the adverse effects of GH.
Estrogen in Adipose Tissue
Men and women have a different distribution of body fat; a gluteal-femoral pattern is seen in women and an abdominal pattern in men. This sexual dimorphism is thought to be due to differences in the sex steroids estrogen and testosterone. Lipoprotein lipase (LPL) activity, indicative of lipid storage, is increased in the gluteal-femoral region of women as compared with men. After menopause, LPL activity is equivalent across all adipose tissue depots, suggesting that estrogen upregulates LPL in a depot-specific fashion.189 In support of this concept, treatment of postmenopausal women with estradiol increased LPL activity in the gluteal-femoral region,190 and this was reversed by the addition of a progestin.191 In vitro in human abdominal subcutaneous adipocytes, low-dose estradiol increased LPL protein and higher-dose estradiol decreased LPL.192 These dose-dependent effects of estradiol in decreasing LPL were also observed in a cross-sectional study193 and after local transdermal application of estradiol.194 In addition to systemic estradiol, the stromal-vascular fraction of adipose tissue is able to convert estrogenic precursors to estrogen vis-à-vis the enzyme aromatase. In men, testosterone, but not the nonaromatizable steroid dihydrotestosterone, increases adipose tissue lipid turnover, suggesting that testosterone acts in adipose tissue via local conversion of testosterone into estrogen by aromatase.195,196 In vitro, estradiol increases the proliferation of stromal-vascular cell cultures of both human197 and rodent preadipocytes.198
Several investigators199–202 have demonstrated estradiol binding and estrogen receptors mRNA in adipose tissue extracts. After cloning of the ER gene, both mRNA and protein for estrogen receptors were subsequently described in adipose tissue.201–203 In human adipose tissue, estrogen receptors was higher in abdominal compared with gluteal femoral adipose tissue, and regional differences in adipose tissue expression of estrogen receptors were described by Pedersen et al.204 However, not all of the effects of estrogen occur in the adipose tissue. By administering estrogen directly into the brain, one can reverse the gain in visceral fat seen with ovariectomy, suggesting that much of the fat patterning attributed to peripheral effects might actually be mediated through hypothalamic signaling.205
In summary, estradiol is an important sex steroid, both for the proliferation of adipocyte precursors and for the regulation of lipid storage in a region-specific fashion.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


