Chapter 16 Ritonavir
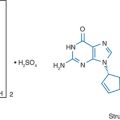
Ritonavir (RTV) is an inhibitor of the protease enzyme encoded by the human immunodeficiency virus (HIV) (Fig. 16-1). The chemical structure of the synthetic peptidomimetic antiviral agent RTV was designed based on the C2 symmetry of this enzyme.1,2 HIV protease cleaves the gag and gag-pol precursor molecules into smaller proteins. Without proper cleavage, the structural proteins of the virion core (p17, p24, p9, p7) and viral enzymes (reverse transcriptase (RT), integrase, protease) cannot be formed adequately, which results in the formation of immature, noninfectious virions.3,4 Early in vitro studies showed equal potency of RTV and zidovudine (ZDV) on a molar basis for several laboratory HIV-1 strains in MT-4 cells and eightfold less activity against an HIV-2 strain, with a median inhibitory concentration (IC50) of 0.045 μmol/L against seven clinical HIV-1 isolates in peripheral blood lymphocytes.1 The affinity of RTV for human aspartic proteases such as renin, pepsin, cathepsin D and E, and gastricin is low; and cytotoxic effects are seen only when RTV is administered in concentrations of more than 500–1000 times those required for antiretroviral activity.1,2
The spectrum of antiviral activity is limited. RTV is active against human retroviruses including HIV-1 and, to a lesser degree, HIV-2.2,3,5
PHARMACOKINETICS
The pharmacokinetics of RTV have been studied in healthy volunteers and HIV-infected adults 18–63 years of age. The pharmacokinetics in adults above the age of 63 and in persons with kidney or liver function disturbances have not been assessed.6 So far the results do not reveal differences in pharmacokinetics between healthy individuals and HIV-infected persons or between individuals of different gender or race.
Following oral administration, RTV is well absorbed, with peak plasma concentrations appearing within 2–4 h.7 The maximal concentration (Cmax) and area under the concentration–time curve (AUC) increased nonlinearly after single doses ranging from 100 to 1000 mg: The mean dose-normalized (per 100 mg) C max and AUC increased from 0.416 μg/mL and 3.480 μg h−1 mL−1 (100 mg) to 1.27 μg/mL and 12.31 μg h−1 mL−1 (1000 mg), respectively. This points to a saturable metabolism.7 In a phase II clinical trial, after 3 weeks of treatment with RTV monotherapy, the Cmax and AUC varied from 5.7 μg/mL and 29.7 μg h−1 mL−1, respectively, with a 300 mg bid dose to 11.2 μg/mL and 60.8 μg h−1 mL−1, respectively, with a 600 mg bid dose.8 After oral dosing of 600 mg of 14C-labeled RTV, 34% of the dose was recovered from the feces as unchanged drug, and virtually all of the RTV in the systemic circulation was unchanged, indicating an oral bioavailability of at least 60% and a small first-pass effect.6
Although the presence of food in the gastrointestinal tract affects the rate and extent of absorption of RTV, it does so only to a moderate degree and is dependent on the dosage form of the drug.6 Administration of 600 mg of RTV as oral solution with food results in a delayed and decreased (∼23%) peak plasma concentration and 7% decreased overall absorption when compared to administration of the same dose in the fasting state. However, the overall absorption of RTV administered as capsules seems to be increased (by ∼15%) when taken simultaneously with a meal.6
Between 98–99% of RTV is bound to plasma proteins; most of it is bound to albumin and α1-acid glycoprotein over a large concentration range (0.01–30 μg/mL). Currently it is not known if RTV crosses the placenta or is excreted in breast milk.6
The exact relation between plasma RTV concentrations and antiretroviral effects has not been determined. In patients receiving RTV 600 mg bid as an oral solution or as capsules, the plasma trough concentrations exceed the concentration of RTV (after adjustment for binding to human plasma proteins) required to inhibit 90% of detectable HIV-1 replication in vitro, the so-called IC90.8
Elimination of RTV is mainly by hepatic metabolism.6,9 In patients receiving 600 mg bid, the systemic clearance was 8.8 L/h, and the renal clearance has been reported to be less than 0.1 L/h. Plasma half-life averages 3–5 h.6,8 In the human liver microsomes, RTV metabolism is mediated by the cytochrome P450 (CYP) subsystems 3A4 and, to a lesser degree, 2D6. As stated above, after administration of 600 mg radio-labeled RTV as an oral solution, 34% of the drug was excreted in the unchanged form, and 86% of the dose was excreted in the feces. In total, 11% of the dose was excreted in the urine, of which only 3.5% was excreted as unchanged drug.6 There are no data available on elimination by hemodialysis or peritoneal dialysis; but given the hepatic metabolism and the high degree of binding to proteins in the plasma, it seems unlikely that substantial amounts of the drug can be eliminated via these means.
DRUG INTERACTIONS
Because RTV has a high affinity for several P450 isoenzymes, the compound exhibits clinically important drug interactions with a wide variety of other drugs. Isoenzymes CYP3A4, CYP2D6, CYP2C9, CYP2D19, and to a lesser degree CYP2A6, CYP1A2, and CYP2E1 are affected by RTV. Co-administration of RTV and drugs that are metabolized by these isoenzymes may result in competition for these enzymes, causing a decrease in metabolism and high plasma levels of the other agents. Conversely, RTV is metabolized by CYP3A4 and CYP2D6,9 and therefore concomitant administration of RTV and drugs that induce these isoenzymes may result in decreased RTV plasma levels. Finally, RTV may increase the activity of glucuronyltransferase, and concomitant administration of RTV and drugs that are directly glucuronidated may result in loss of activity of these drugs.6
The manufacturer of RTV issues a regularly updated list of compounds that should not be used or be used with caution in conjunction with RTV. Pharmacokinetic interaction studies with RTV are available for some drugs (e.g., the antifungal fluconazole10), but for many others the recommendations are based on theoretical considerations. Table 16-1 lists recommendations for concomitant use of several drugs with RTV; this list is based on information from the manufacturer6 and is by no means exhaustive.
Table 16-1 Drug Interactions with RTV
| Drug Administered with RTV | Recommendation | Comments |
|---|---|---|
| Antifungals | ||
| Fluconazole | No adjustment | Only small increase in Cmax and AUC10 |
| Itraconazole | Caution | |
| Ketoconazole | Caution | |
| Antimalarial and Antiprotozoals Agents | ||
| Quinine | Caution | Threefold increase in AUC may be expected |
| Proguanil | No adjustment | |
| Chloroquine | Caution | |
| Primaquine | Caution | |
| Pyrimethamine | Caution | |
| Atovaquone | Caution | Possible decreased AUC as a result of enhanced glucuronidation |
| Antimycobacterials | ||
| Rifabutin | Contraindicated | Large increase in AUC of rifabutin, and especially of its 25-O-desacetyl metabolite6 |
| Rifampin | Caution | Threefold increase in AUC of rifampin, but large reduction in AUC of RTV6 |
| Ethambutol | No adjustment | Anecdotal data only; no proper evaluation done |
| Macrolide Antibiotics | ||
| Clarithromycin | Depends on renal function | No adjustment in patients with normal renal function; 50% dose reduction when creatinine clearance is 30–60 mL/min, 75% dose reduction when <30 mL/min |
| Erythromycin | Caution | Threefold increase in AUC of erythromycin to be expected |
| Other Antiinfectives | ||
| Cotrimoxazole | No adjustment | 20% decrease in AUC of sulfamethoxazole and 20% increase in AUC of trimethoprim expected |
| Metronidazole | Discouraged | Disulfiram-like reaction possible (RTV oral solution and capsules contain alcohol) |
| Anticoagulants | ||
| Warfarin | Caution | Large increase in AUC of R-enantiomer and moderate decrease in AUC of S-enantiomer: monitor prothrombin time carefully |
| Antihistaminics | ||
| Astemizole, terfenadine | Contraindicated | Very large increase in AUC expected |
| Antineoplastics | ||
| Group I: etoposide, paclitaxel, tamoxifen, vincristine, vinblastine | Caution | These agents are metabolized by CYP3A4, so threefold or larger increase in AUC can be expected: monitor therapeutic and toxic effects carefully |
| Group II: cyclophosphamide, daunorubicin, doxorubicin | Caution | All these agents are metabolized by unidentified CYP isoenzymes, so changes in AUC are unpredictable: monitor therapeutic and toxic effects carefully |
| Cardiovascular Agents | ||
| Group I: amiodarone, bepridil, encainide, flecainide, propafenone, quinidine | Contraindicated | |
| Group II: diltiazem, disopyramide, felodipine, isradipine, lidocaine, nicardipine, nifedipine, verapamil | Caution | All are metabolized by CYP3A4, so threefold or larger increase in AUC is expected |
| Group III: mexiletine, metoprolol, pindolol, propranolol, timolol | Caution | All are metabolized by CYP2C9/19 isoenzymes; moderate increase or decrease in AUC is to be expected |
| Group IV: acebutolol, digoxin, prazocin, tocainide | Caution | Metabolized by unidentified P450 isoenzymes; no data available |
| Antilipemic Agents | ||
| Lovastatin, pravastatin fluvastatin, gemfibrozil | Caution | Metabolized by CYP3A4 |
| Simvastatin | Contraindicated | Extensively metabolized by P450 isoenzymes |
| Clofibrate | Monitor effect | Metabolized by glucuronidation; might result in decrease of AUC |
| Central Nervous System Agents | ||
| Pyroxicam | Contraindicated | |
| NSAIDs | ||
| Diclofenac, ibuprofen, indomethacin | No adjustment | Only moderate increase in AUC expected |
| Ketoprofen, naproxen | Monitor effect | Metabolized by glucuronidation; might result in decrease of AUC |
| Opiate agonists | ||
| Meperidine, propoxyphene | Contraindicated | |
| Alfentanyl, fentanyl | Caution | Threefold or larger increase in AUC expected |
| Methadone | Caution | Metabolized by unidentified P450 isoenzymes, so increase in AUC possible; also metabolized by glucuronidation, which might result in lowered AUC; net effect unknown |
| Codeine, morphine | Monitor effect | Metabolized by glucuronidation; might result in decrease of AUC |
| Anticonvulsants | ||
| Carbamazepine | Caution | Threefold or larger increase in AUC; decrease of RTV AUC |
| Phenobarbital | Caution | Metabolized by unidentified P450 isoenzymes, so increase in AUC possible; decrease in RTV AUC possible |
| Phenytoin | Caution | Moderate increase or decrease in AUC expected; decrease in RTV AUC possible |
| Sedatives and hypnotics | ||
| Alprazolam, clorazepate, diazepam, estazolam, flurazepam, midazolam, triazolam, zolpidem Lorazepam, oxazepam, propofol, temazepam | Contraindicated | |
| Caution | Metabolized by glucuronidation; might result in decrease of AUC | |
| Gastrointestinal Agents | ||
| Dronabinol, ondansetron | Caution | Metabolized by CYP3A4: threefold or larger increase in AUC |
| Lansoprazole, omeprazole | No adjustment | Moderate increase or decrease in AUC to be expected |
| Promethazine | Caution | Metabolized by unidentified P450 isoenzymes; increase in AUC possible |
| Corticosteroids | ||
| Prednisone | Caution | Metabolized by CYP3A4: threefold or larger increase in AUC |
| Dexamethasone | Caution | Metabolized by CYP3A4: threefold or larger increase in AUC; also, induction of CYP3A4 possible, with resulting increase in RTV clearance and lowering of plasma RTV levels |
| Estrogens | ||
| Ethinyl estradiol | Increase of dose or alternative contraceptive method | RTV 500 mg bid decreased Cmax and AUC 32% and 40%, respectively6 |
AUC, area under the time – concentration curve; NSAIDs, nonsteroidal antiinflammatory drugs.
The strong affinity of RTV for some P450 isoenzymes may also influence the pharmacokinetics of several other antiretroviral agents when given in combination with RTV. Concomitant administration of RTV (600 mg bid) and nucleoside analog reverse transcriptase inhibitors (NRTIs) such as didanosine (200 mg bid) or zidovudine (ZDV) (200 mg tid) over 4 days decreased the Cmax by 16% and 27%, and decreased the AUC by 13% and 25%, respectively.11 The manufacturer does not advocate dose adjustments of either of these antiretroviral agents.
Co-administration of RTV and the non-nucleoside reverse transcriptase inhibitor (NNRTI) nevirapine resulted in a mean nonsignificant 11% decrease in RTV AUC in 12 HIV-infected individuals12; the change in nevirapine pharmacokinetics also was not significant. Co-administration with another NNRTI, delavirdine, likewise had no clinically relevant effects on the steady-state pharmacokinetics of either RTV or delavirdine in a study using RTV at a reduced dose.13
In contrast to the co-administration of RTV and RT inhibitors, the simultaneous use of RTV and other protease inhibitors (PIs) leads to important pharmacokinetic interactions. RTV causes a highly significant increase in both the Cmax and the AUC of saquinavir (SQV) when the two drugs are administered simultaneously. This information is useful because the oral bioavailability of SQV is poor as a result of, among other causes, an extensive first-pass effect.14 Whereas the standard dose of SQV in combination with NRTIs is 600 mg tid, when SQV is co-administered with RTV the AUC of SQV is so greatly increased that even 400 mg bid dosing results in a 20–40-fold increase without much effect on RTV pharma-cokinetics.15,16 The soft gel capsule formulation of SQV is administered at a dosage of 1200 mg tid. However, when used with RTV, the dose administered is the same as that recommended for the older formulation of SQV: 400–600 mg bid.
The pharmacokinetics of the PI indinavir (IND) are heavily influenced by co-administration of RTV as well. In healthy volunteers, RTV increased the AUC and Cmax of IND by 480% and 110%, respectively; even with a reduced, twice-daily dosing regimen of IND without the usual administration of the drug in the fasting state, the combination of RTV and IND yielded a comparable AUC, with higher trough and slightly lower peak levels of IND compared to the standard three-times-a-day regimen. No effects on RTV pharmacokinetics were observed.17 RTV also increased the AUC of the PI nelfinavir by 150%, without affecting the AUC of RTV.18,19
CLINICAL TRIALS
RTV Used As a Sole PI
Ritonavir monotherapy has been investigated in two phase I/II trials.8,20 Danner et al in a double-blind, randomized, placebo-controlled trial, studied 84 HIV-positive patients who had no prior exposure to a PI and at least 50 CD4+ T lymphocytes per cubic microliter. They were assigned to one of four RTV dosing regimens (300, 400, 500, or 600 mg bid) or placebo for 4 weeks, then the placebo recipients were again randomized to one of the RTV regimens.8 During the first 4 weeks, increases in CD4+ T lymphocytes and decreases in plasma HIV RNA were similar among the four RTV groups and significantly different from those in the placebo groups. Thereafter, however, a return to baseline values was noted after 16 weeks among the three lower dosage groups. In the highest dosage group, the HIV RNA load also partially increased after the nadir was reached at 4–8 weeks of treatment (1.2 log10 copies/mL decrease). After 32 weeks; among the patients in the 600 mg bid dosage group, the median increase from baseline in the CD4+ T-lymphocyte count was 230 cells/mL, and the mean decrease in HIV RNA was 0.81 log10 copies/mL. However, this last parameter was assessed with the first-generation Chiron branched-chain DNA (bDNA) assay, which had a lower limit of detection of 10 000 RNA copies/mL.21 When a more sensitive assay was utilized, decreases in the HIV RNA load turned out to be larger; applying the Roche polymerase chain reaction (PCR) assay22 in the two highest dose groups, a mean maximal decrease of 1.91 log10 copies/mL was found, in contrast to 1.1 log10 copies/mL when measured with the bDNA assay. Adverse events included nausea, circumoral paresthesias, and elevated transaminase and triglyceride levels. Ten withdrawals from the study were judged to be RTV related.
In another phase I/II trial, Markowitz et al studied three- and four-times-a-day dosing regimens of RTV in 62 HIV-infected patients.20 As in the prior study, a clear increase in CD4+ T-lymphocyte count and a decreased plasma HIV RNA load were found over 12 weeks, although the responses in these dosage groups were slightly inferior to those in the 600 mg bid regimen mentioned in the previous study. In this study pharmacokinetics were studied early (days 4–11) and later (days 22–29) during RTV administration. In all dosing groups the Cmax, minimal concentration, and AUC decreased considerably during the period between the two time points, which is in agreement with preclinical studies suggesting that RTV induces its own metabolism.1 The spectrum of adverse events was not different from that seen in the first phase I/II study. Of the 62 patients, three withdrew because of adverse events thought to be related to RTV administration.20
Use in Antiretroviral-Naive HIV-Infected Patients
Notermans et al23 treated 33 antiretroviral-naive HIV-infected patients (31 men, two women) with a CD4+ T-lymphocyte count of more than 50 cells/mL and a plasma HIV RNA load of more than 30 000 copies/mL (Roche Amplicor) with RTV, ZDV, and lamivudine (3TC) (Table 16-2). The patients were randomized into two groups. Group I started with all three antiretroviral agents simultaneously, and group II started with RTV monotherapy for 3 weeks; ZDV and 3TC were then added (this design was chosen to investigate whether quick development of resistance against the nucleoside analogs, especially against 3TC, could be prevented by prior lowering of the HIV replication rate by a PI). A tonsillar biopsy was performed at baseline and after 2 days, 22 days, and 6 months of therapy to study changes in the HIV RNA load and lymphocyte subsets in lymphoid tissues.24 Group I had a somewhat higher CD4+ T-lymphocyte count than group II (177 vs 134 cells/mL), but there were no differences in the baseline HIV RNA load (mean 5.3 log10 copies/mL) or in the Centers for Disease Control and Prevention (CDC) class of clinical HIV infection. Protease and RT gene sequencing at baseline showed only one case with a 41 and 215 RT codon change (known to confer resistance to ZDV) pretherapy, and in six cases two protease gene mutations (36, 71, or both) not known to be associated with resistance against RTV were observed. During treatment there was a quick initial rise in CD4+ T lymphocytes, with a more gradual increase with a nonsignificant, slightly better result after 24 weeks in group I (an increase in CD4+ T lymphocytes of 180 versus 99 cells/mL in group II). A protease gene codon 82 mutation (known to confer resistance to RTV) developed in only one patient on therapy who did not comply with the regimen. No new RT gene mutations developed in any of the patients. The plasma HIV RNA load showed an initial rapid decline during the first 3 weeks, followed by a slower further decline, reaching its nadir of almost 3.0 log10 copies/mL at week 16 (Fig. 16-2).23 The decreased plasma viral load was mirrored in the lymphoid tissue HIV RNA load. Both the amount of HIV trapped on the surface of follicular dendritic cells and the number of actively HIV-producing mononuclear cells decreased sharply, even after 2 days of therapy, and reached a decrease of more than 3.4 and more than 2.3 log units per gram of lymphoid tissue, respectively.24 There were many adverse events, mainly gastrointestinal in nature, prompting eight of 33 patients to withdraw from the study.
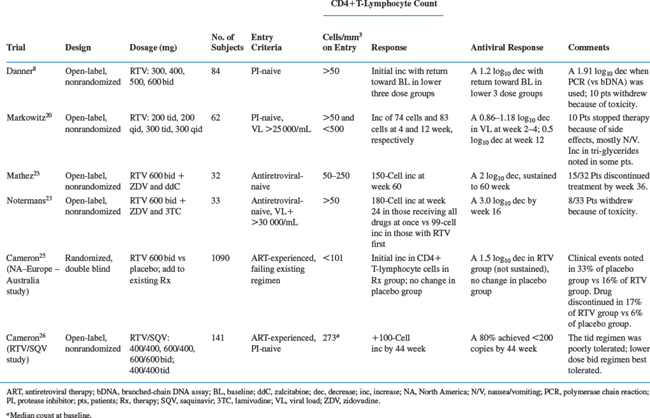
In contrast to the phase I/II studies in which RTV was administered as monotherapy, in this study the excellent HIV RNA response was maintained for at least 6 months. For example, in the RTV/ZDV/3TC study, 95% of the patients who were still on treatment after 52 weeks had no detectable plasma HIV RNA load in the Roche Amplicor assay (S.A. Danner, unpublished results).
Saimot et al27 evaluated the efficacy and safety of RTV in combination with stavudine (d4T) and didanosine as first-line antiretroviral treatment in an open-label study in 33 patients with 50–350 CD4+ T lymphocytes per mL. After 2 weeks of RTV induction the nucleoside analogs were added. At 12 weeks of treatment, both infectious blood cells (cells per 107 peripheral blood mononuclear cells (PBMCs)) and plasma HIV RNA (log10 copies/mL) had decreased significantly, with reductions of 2.06 and 2.14, respectively. Six patients (18%) discontinued therapy during the first 5 weeks because of adverse events.27
Markowitz et al treated patients presenting with primary HIV infection with ZDV/3TC/RTV or IND.28 The time from the onset of symptoms to the start of treatment was 55 days. In the RTV-treated group, seven of 12 patients maintained their regimen for 10–16 months; in the IND group, 11 of 12 patients remained on therapy for 4–9 months. All subjects achieved undetectable levels of plasma HIV RNA (modified Chiron 2.0 assay, with a lower limit of detection of 100 copies/mL), and quantitative cultures went below the threshold of detection (0.1 TCID50/106 PBMC) within months in all compliant subjects. Moreover, PBMCs isolated from semen collected from selected subjects did not express spliced or unspliced HIV-1 mRNA. No signs of viral replication could be found in lymphoid biopsy material from the sigmoid colon. The CD4+ T-lymphocyte count and the CD4/CD8 ratio increased, and reductions in env- and gag-specific antibody titers were noted.
Use in Antiretroviral-Experienced HIV-Infected Patients
In 1994, the efficacy and safety of RTV were evaluated in a 68-center North American–European–Australian study in patients with clinically advanced disease for whom there were no other treatment options. The study enrolled 1090 HIV-infected persons with a CD4+ T-lymphocyte count of less than 101 cells/mL and at least 9 months of prior antiretroviral treatment. Either RTV or placebo was added in a double-blind fashion to their existing antiretroviral medication.25 Crossover to open RTV for any acquired immunodeficiency syndrome (AIDS) outcome was permitted after 4 months. The patients had far-advanced HIV infection, with baseline median CD4+ T-lymphocyte counts of 18 and 22 cells/mL for the RTV and placebo groups, respectively. The patients had been treated with antiretroviral drugs for a mean of 2.5 years, with an average of more than 2.5 drugs. Drug discontinuation because of adverse events occurred in 17% of the RTV group and 6% of the placebo group, mostly because of gastrointestinal symptoms during the first few weeks of the study. In the placebo group, no plasma HIV RNA-lowering effect was observed. In the RTV group, there was an initial decrease of ±1.5 log10 copies/mL that was not sustained after 6 months. The study was designed with clinical endpoints (death or occurrence of a new AIDS-defining event). Death or an AIDS-defining event occurred in 85 (15.7%) versus 181 (33.1%) patients in the RTV and placebo groups, respectively (P < 0.001, hazard ratio 0.44%, 95% confidence interval (CI) 0.34–0.56). Although the study was not powered to detect the presumed difference in deaths, it turned out that the number of deaths differed significantly between the two groups. After a median 6.1 months of follow-up, 26 (4.8%) patients died in the RTV group versus 46 (8.4%) patients in the placebo group, a highly significant difference (P = 0.02, hazard ratio 0.57, 95% CI 0.35–0.91).25
Given the long period of prior antiretroviral treatment, it can be assumed that many patients in this study must have been nucleoside analog resistant. As a result, these patients were treated with RTV monotherapy. This is in agreement with the transient nature of the plasma HIV RNA decrease, pointing to rapid development of viral resistance against the drug. Currently, with more antiretroviral agents available, monotherapy with RTV is no longer considered acceptable even in patients with advanced disease. Because of expected rapid loss of viral susceptibility to the drug, a favorable clinical result of such a treatment must be judged remarkable. This study was instrumental in securing approval of the US Food and Drug Administration’s (FDA’s) New Drug Application for RTV.
Use of RTV in Pharmacologic “Boosting” of Other HIV PIs
RTV is not only an effective HIV PI, it is also one of the most powerful inhibitors of the P450 cytochrome system, especially of the isoenzyme CYP 3A4.15,16 Because all other licensed HIV-PIs are largely metabolized by CYP3A4, RTV can be used to “boost” the efficacy of these compounds by enhancing their biologic availability.
Because RTV at its full, HIV replication suppressing dose (600 mg bid) has so many side effects, and the improvement of pharmacokinetics of other co-administered PIs is so impressive, the drug is currently almost exclusively used as a “booster” for other PI’s. Substantial data are available on the use of the compound in this way. RTV has been combined with SQV, IND, nelfinavir, lopinavir (LPV) (co-formulated with RTV in one capsule/tablet/liquid), amprenavir, fosamprenavir, and atazanavir. Most of the efficacy results of such trials are discussed in the respective chapters devoted to these compounds. Here a short summary is provided, mainly focusing on pharmacokinetics. Extensive reviews are written by Cooper et al29 and by Zeldin and Petruschk.30
SQV/RTV
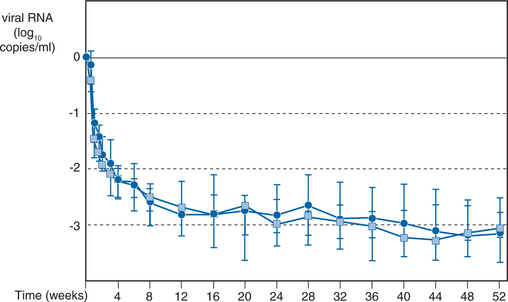
The combination RTV/SQV has been studied in a dose-finding clinical trial.26,31 A total of 141 patients with a median baseline CD4+ T-lymphocyte count of 273 cells/mL and a median plasma HIV RNA load of 4.63 log10 copies/mL who discontinued their RT inhibitor therapy, took part in a multicenter, randomized, open-label study. The patients were randomized into four groups with various two- and three-times-a-day regimens: 400 mg RTV/400 mg SQV bid; 600 mg RTV/400 mg SQV bid; 400 mg RTV/400 mg SQV tid, and 600 mg RTV/600 mg SQV bid. All four dosage groups showed an excellent virologic and immunologic response: After 44 weeks, a median increase in CD4+ T lymphocytes to more than 100 cells/mL and a plasma RNA load decrease to less than 200 copies/mL were noted in more than 80% of the patients (intention-to-treat analysis). After 12 months, 90% of the patients on treatment still had low plasma RNA load values (Fig. 16-3). The most important difference between the four groups was tolerability: In the lowest-dose group (35 patients), few adverse events were noted, with only one discontinuation resulting from an adverse event. In seven patients who responded incompletely, d4T/3TC therapy was added at week 12, after which they also reached less than 200 copies/mL HIV RNA load.26 Interestingly, the most important predictor of the duration of the viral response was the nadir in plasma viral load that was achieved. An HIV RNA load below 200 copies/mL at 6 months was predicted at 3 months by a level of less than 1000 copies/mL (92% positive predictive value (PPV), 75% negative predictive value (NPV); P = 0.005) and a level of 200 copies/mL (93% PPV, 33% NPV; P < 0.05).32 This observation has been confirmed in studies with other antiretroviral drugs. For example, in the INCAS study with ZDV, didanosine (ddI), and nevirapine, the multivariate model revealed that neither the baseline plasma viral load, the viral response, the baseline CD4+ T-lymphocyte count, nor the CD4+ T lymphocyte response was independently associated with the duration of the response; rather, the nadir of the plasma viral load was highly predictive and accounted for 78% of the treatment difference in duration of suppression.33 The favorable antiviral effect in the RTV/SQV study was even seen after 3 and 4 years of treatment, with a high virologic response in an intention-to-treat analysis.34 At the beginning of the fourth year, 48 of 84 patients were still being treated successfully with only RTV/SQV.35



Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


