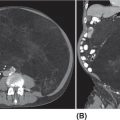
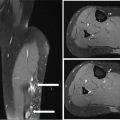
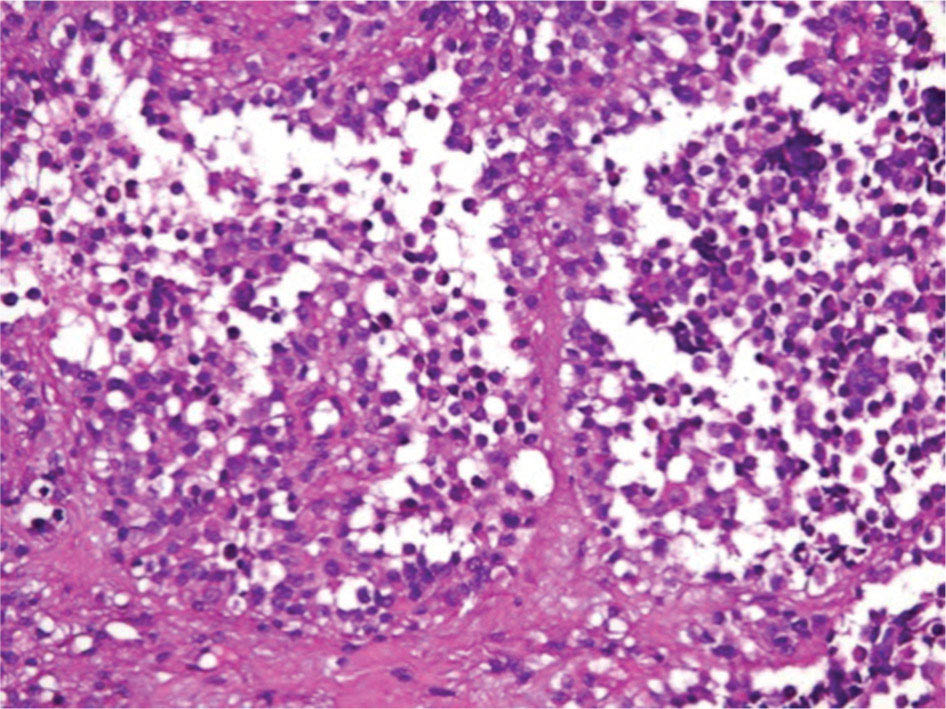
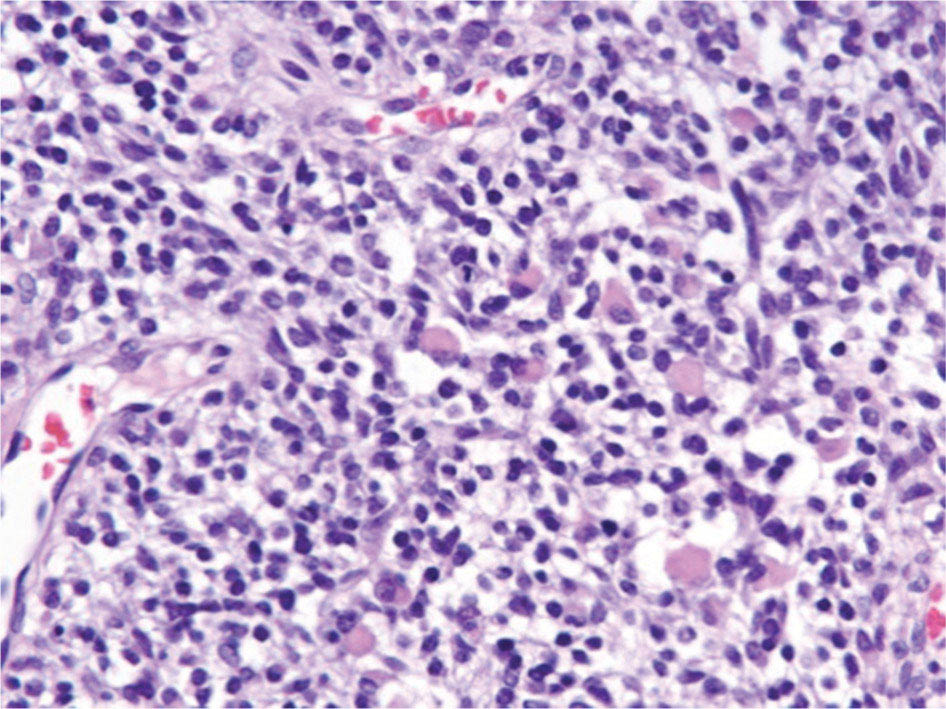
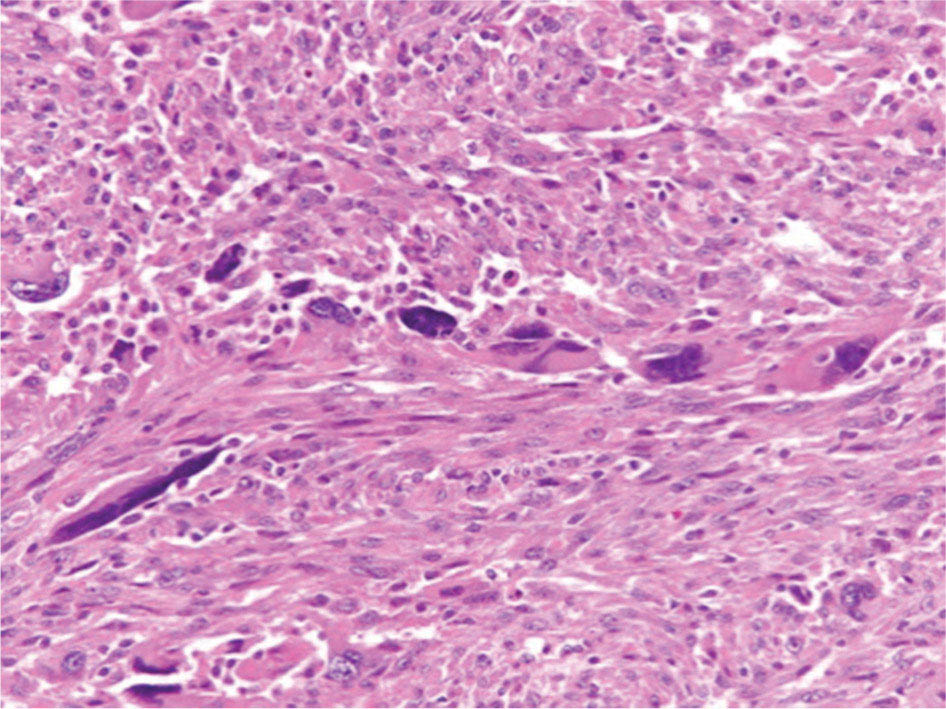
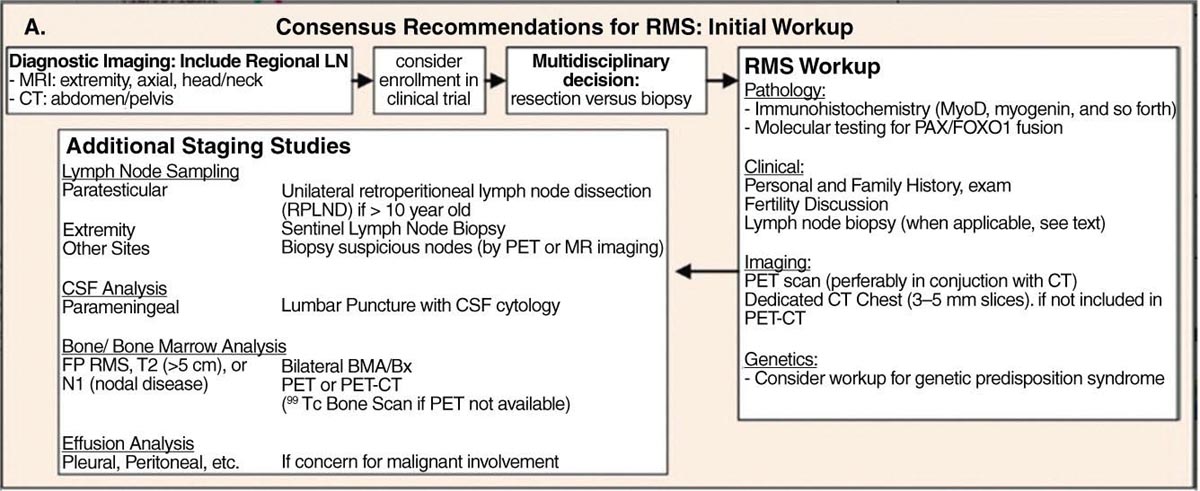
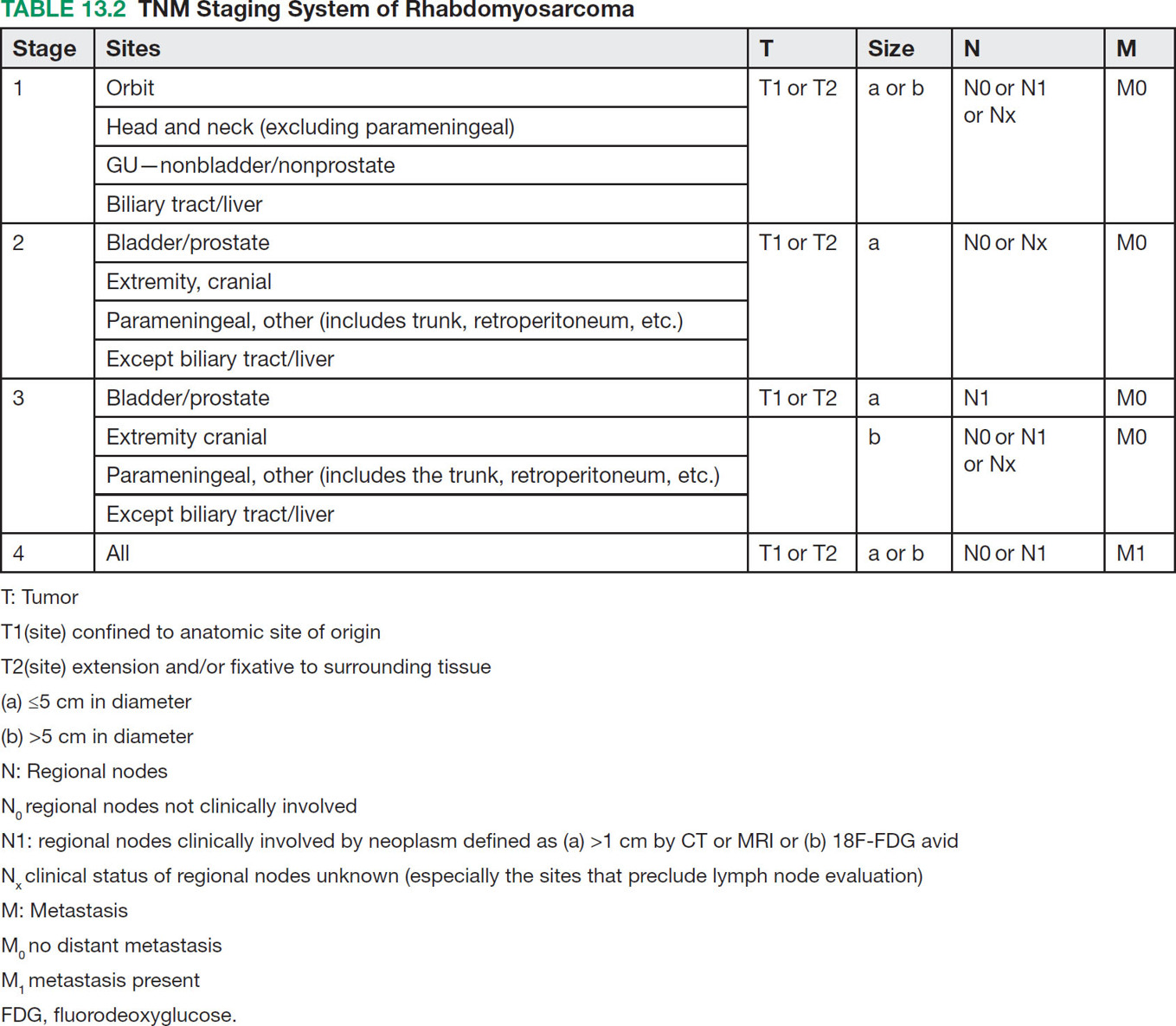
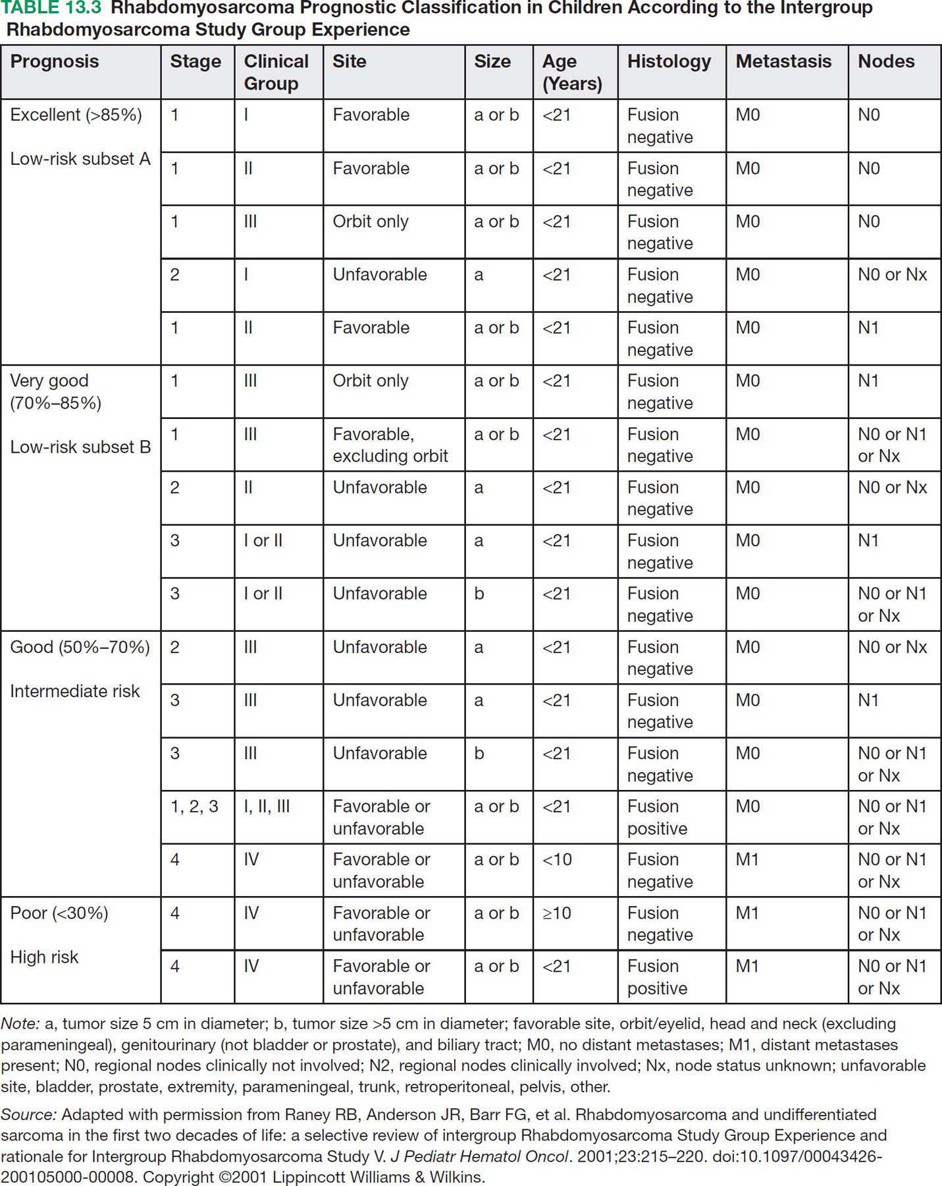
17113 Rhabdomyosarcoma Rhabdomyosarcoma (RMS) is a soft tissue sarcoma composed of small round cells and spindle cells of unknown origin mimicking the striated aspect of skeletal muscle. RMS is traditionally classified into embryonal, alveolar, and pleomorphic subtypes. The spindle cell/sclerosing subtype, once considered a variant of embryonal subtype, is now a distinct fourth subtype according to the 2015 World Health Organization classification. Being the most common soft tissue sarcoma and the third most common extracranial solid tumor after neuroblastoma and Wilms tumor in children, RMS is generally considered a pediatric tumor. With the combined use of chemotherapy, radiation therapy, and surgery, 80% of patients with localized disease remain alive at 5 years. Management of adult RMS is still extrapolated from pediatric protocols, despite several studies showing distinct biologic and clinical disease patterns. This chapter discusses the diagnostic approach, pathology, staging, prognostic factors, and treatment of RMS. Rhabdomyosarcoma, embryonal, alveolar, FOXO1 protein, sarcoma, adult, pediatric, chemotherapy protocols, multidisciplinary management, young adult chemotherapy, neuroblastoma, radiation therapy, rhabdomyosarcoma, surgery, Wilms tumor Drug Therapy, General Surgery, Neuroblastoma, Radiotherapy, Rhabdomyosarcoma, Wilms Tumor INTRODUCTION Rhabdomyosarcoma (RMS) is a soft tissue sarcoma composed of small round cells and spindle cells of unknown origin mimicking the striated aspect of skeletal muscle, thus the Greek prefix “rhabdo” meaning rod shape. RMS is traditionally classified into embryonal, alveolar, and pleomorphic subtypes. The spindle/sclerosing subtype, once considered a variant of embryonal subtype, is now a distinct fourth subtype according to the 2015 World Health Organization (WHO) classification. Being the most common soft tissue sarcoma and the third most common extracranial solid tumor after neuroblastoma and Wilms tumor in children, RMS is generally considered a pediatric tumor.1 With the combined use of chemotherapy, radiation therapy (RT), and surgery, 80% of patients with localized disease remain alive at 5 years. Conversely, the 5-year overall survival (OS) rates seldom exceed 30% in patients with metastatic disease.2 On the other hand, 40% of RMS cases occur in adults. Management of adult RMS is still extrapolated from pediatric protocols, despite several studies showing distinct biologic and clinical disease patterns. More studies are needed in this specific patient population in order to improve the outcomes of this rare and heterogeneous disease. EPIDEMIOLOGY Incidence RMS is a rare tumor that accounts for 4% of childhood malignancies and about 3% of all adult soft tissue sarcomas in the United States. According to the Surveillance, Epidemiology, and End Results (SEER) database, 350 new cases are diagnosed each year in the United States and the annual incidence of RMS in children and adults younger than 20 years of age is around 4.3 cases per 1 million. The French Sarcoma Group published one of the largest prospective cohorts performed in a multicenter setting at a national level for adult RMS. The estimated incidence of adult RMS was around 0.9 case per million per year.3 Half the cases occur in children under the age of 10, while 40% of them affect adults. Incidence of pediatric RMS is highest between the ages of 2 and 6 years, and 10 and 18 years. Adult RMS usually occurs between the third and seventh decades of life. Embryonal RMS (E-RMS) affects approximately four cases per 1 million children younger than 4 years of age. It is less frequent in adolescents and very rare in adults. Incidence of alveolar RMS (A-RMS) is around one per 1 million children and adolescents. Pleomorphic RMS (P-RMS) occurs in <2% of children, but is the second most frequent entity in the adult population (19%). There is a slight male predominance in children with a male to female ratio between 1.3:1 and 1.5:1 that varies according to the histologic subtype (E-RMS being more common in males, while A-RMS is gender independent) and tumor location (predominance of genitourinary [GU] tract and orbital tumors in males and females, respectively). On the other hand, the incidence of RMS is equivalent between male and female adults. In addition to being gender dependent, tumor location also varies with age. Children more often present with GU tract, orbital, and head and neck tumors that are almost always of the embryonal type. Adolescents and adults experience more extremity, truncal, and paratesticular tumors that are frequently of the alveolar type. Incidence of RMS is slightly higher in black patients between 15 and 19 years of age, compared to white patients. It is lowest in the Asian population.4 No geographic distribution has been reported. 172Risk Factors There is a lack of solid data concerning risk factors. Some studies suggested a higher incidence of RMS in patients previously exposed to alkylating agents or to radiation in utero, those with accelerated in utero growth, children from families with a low socioeconomic status, those who received antibiotics soon after birth, and those whose parents used recreational drugs (marijuana or cocaine) during pregnancy.5–7 Other studies suggest a protective role for higher birth order, immunizations, and immune-related factors (allergies, atopy, day-care attendance, breastfeeding for 12 or more months).8–10 Inherited Syndromes Most cases of RMS are sporadic; however, Zhang et al. identified germline pathogenic or probably pathogenic mutations by next-generation sequencing in 7% of children with RMS.11 In another study by Ruymann et al., congenital abnormalities were found in 37 of 115 (32%) children and adolescents autopsied with RMS. These patients also had an increased incidence of congenital malformations affecting the central nervous system (CNS) or GU, cardiovascular (CV), and gastrointestinal (GI) systems.12 The following hereditary syndromes have a higher incidence of RMS: • Li–Fraumeni syndrome: Inactivating germline mutations of the p53 tumor suppressor genes predispose patients to a spectrum of cancers, including soft tissue sarcomas.13 Incidence of RMS in this setting may be greatest among children under 3 years of age, who should therefore be screened for germline p53 mutations.14 In addition, patients with Li–Fraumeni syndrome usually develop anaplastic forms of RMS.15 • Beckwith–Wiedemann syndrome: This hereditary disease shares the same chromosomal abnormalities (11p15, the locus for the IGF-2 gene) with certain pediatric tumors such as RMS, hepatoblastoma, Wilms tumor, and adrenocortical carcinoma.16 • Costello syndrome: In addition to postnatal growth retardation, macrocephaly, coarse facies, loose skin, nonprogressive cardiomyopathy, developmental delay, and papillomas, the presence of a mutation in the HRAS proto-oncogene predisposes patients to several tumors including RMS, epithelioma, bladder carcinoma, and vestibular schwannoma.17–22 • Neurofibromatosis: Incidence of RMS is 20 times higher compared with the general population.23 • DICER1 syndrome: In this autosomal dominant, pleiotropic, tumor-predisposition disorder resulting from a germline DICER1 gene mutation, there is an increased risk for GU E-RMS.24,25 • Rubinstein–Taybi syndrome: predisposes to neural and developmental tumors including nasopharyngeal RMS.26,27 • Gorlin basal cell nevus syndrome: There are several case reports of RMS associated with this syndrome, a rare autosomal dominant, inherited disorder that is characterized by development of basal cell carcinomas from a young age.28,29 CLINICAL PRESENTATION RMS presents with variable signs and symptoms according to tumor location, patient age, and presence or absence of metastases. While a painless enlarging mass is easily palpated in the head and neck or the limbs, diagnosis of parameningeal or retroperitoneal RMS can be challenging. Head and Neck Primary RMS This is the most frequent primary tumor site (35%–40% of cases). Tumors can arise in the orbit (25%) resulting in proptosis or diplopia, in parameningeal sites (middle ear, nasal cavity, paranasal sinuses, nasopharynx, and infratemporal fossa; 50%) resulting in nasal obstruction or cranial nerve palsies, as well as in other head and neck locations (25%).30 E-RMS predominates and lymph node (LN) involvement is infrequent.31 GU Tract Twenty-five percent of RMS occurs in the GU tract. Embryonal subtype is predominant with specific occurrence of the botryoid variant. Bladder tumors cause hematuria and urinary obstruction. Prostate tumors present as large pelvic masses resulting in obstructive and irritative urinary symptoms, as well as constipation and bowel obstruction. Vaginal tumors may produce a mucosanguineous discharge in young girls, along with a protruding polypoid mass. Older girls present with cervical and uterine tumors. A painless scrotal or inguinal mass may be reported in boys with paratesticular RMS.32 More than half of these boys older than 10 years of age have nodal involvement.33 173Extremities Twenty percent of RMS cases present as a bulging, nontraumatic, nontender mass in the extremities, with or without skin erythema. They mainly occur in adolescents, spread to LNs (50%) and along fascial planes, and are of the alveolar subtype (50%–75%). Metastatic Disease Metastatic disease is identified in <25% of patients at diagnosis. The most frequent metastatic site is the lung (50%). Patients with metastatic disease to the bones (30%) may present with skeletal-related events. Bone marrow involvement occurs in 30% of cases and results in cytopenias leading to a false diagnosis of leukemia. Other metastatic sites include the omentum (16%) and the pleura (13%). Visceral and brain metastases are rare.34–37 DIAGNOSTIC APPROACH Biopsy In patients with a suspicious mass, an open or core-needle biopsy is needed to establish the diagnosis. Whenever there is a suspicion for sarcoma, the patient should be referred to an expert center and the case displayed at the multidisciplinary tumor board. A surgical biopsy should be performed by an experienced sarcoma surgeon to prevent dissemination of tumor cells. An alternative to this method is a 14-gauge core-needle biopsy performed by an experienced interventional radiologist. The diagnostic material should be reviewed by a pathologist with an expertise in this domain, which ensures proper tissue processing and allows for special cytogenetic and molecular studies necessary for diagnosis and classification. Imaging Workup Imaging of the primary tumor site varies according to the tumor location and may include the following: • Plain x-ray studies to identify calcifications and bone involvement of the primary tumor • CT scan to serve as a baseline for future evaluations and to assess for bone erosions • MRI, which evaluates local extension more accurately than CT scan in orbital, paraspinal, and parameningeal locations Once the pathologic diagnosis of RMS is established, the following imaging exams should be performed to rule out metastatic disease: • CT scan of the chest to identify secondary lung lesions • CT scan or ultrasound of the liver in patients with abdominal and pelvic primary tumors • 99Tc bone scan to identify bone metastases • Spinal and pelvic MRI to search for bone marrow involvement and guide subsequent biopsies • 18-Fluorodeoxyglucose (18-FDG) PET-CT scan to search for regional nodal involvement as well as metastatic disease, especially to the bone; also to assess early response to chemotherapy • Brain MRI in patients with neurologic symptoms Laboratory Workup Laboratory studies may reveal abnormalities in favor of metastatic diseases and are necessary to assess the patient’s condition before starting treatment. They include the following: • Complete blood count (CBC) to search for inflammatory anemia or other cytopenia • Renal and liver function exams • Albumin: to evaluate the nutritional status • Urinalysis: to detect hematuria in RMS of the GU tract Other Diagnostic Exams Cooperative group studies often require a full metastatic workup, namely a CT of the chest, a bone scan, and bilateral bone marrow aspiration and biopsy to rule out marrow involvement. However, because the overall incidence of metastatic disease is low, a risk-oriented rather than disease-oriented staging strategy is currently being adopted. In fact, the most important factors that predict metastatic disease are nodal involvement, tumor invasiveness (>5 cm), and tumor histology (fusion-positive [FP] disease). 174Weiss et al., from the Children’s Oncology Group (COG) soft tissue sarcoma committee, showed that the risk of bone and bone marrow metastases is <1% in a retrospective analysis of 955 fusion-negative (FN) RMS patients without nodal or lung involvement.38 Therefore, in selected children (tumors <5 cm, FN RMS, no evidence of nodal disease), omission of bone marrow biopsy is acceptable. On the other hand, lumbar puncture and cerebrospinal fluid analysis could be solely performed in parameningeal disease. Despite the data mentioned above, lumbar puncture for cerebrospinal fluid cytology in parameningeal tumors and bone marrow aspiration and biopsy remain “mandatory” in pediatric RMS. However, this is not the case in adult patients. These explorations are limited to symptomatic adult patients with suspicion of CNS or marrow involvement. Cardiac ultrasound or nuclear medicine test is recommended before initiating chemotherapy with anthracyclines in order to establish a baseline ejection fraction. Genetic counseling should be considered in younger patients with anaplastic features at diagnosis or in those with a family history of malignant disease. PATHOLOGY Morphologic analysis of fixed, paraffin-embedded biopsy tissue samples establishes the diagnosis of RMS which is characterized by the presence of muscle differentiation features. The latter is further classified into embryonal, alveolar, and pleomorphic subtypes. Cytologic material is generally insufficient to confirm diagnosis.39 Immunohistochemistry (IHC) confirms the diagnosis by using specific antibodies that detect antigens involved in myogenic differentiation. Molecular biology using fluorescent in situ hybridization (FISH) as well as reverse transcriptase-polymerase chain reaction (RT-PCR) assays when FISH is unavailable or uninformative is a complementary diagnostic tool whenever morphologic diagnosis is difficult to obtain. In addition, it may detect chromosomal and molecular abnormalities with prognostic implications. Currently, FISH, next-generation sequencing, or RT-PCR for FOXO1 rearrangement is performed on all RMS tumors, regardless of the histology. Morphology RMS is composed of small round blue cells that resemble other pediatric tumors such as lymphoma, small cell osteosarcoma, mesenchymal chondrosarcoma, and Ewing sarcoma. Identification of rhabdomyoblasts or cross striations that are characteristic of skeletal muscle on light microscopy is necessary to classify a tumor as RMS. Tumor architecture helps determine the histologic subtype: while small round cells grow in cohesive sheets in E-RMS, they are typically detached in A-RMS. In the botryoid variant of E-RMS, there is a typical cambium layer composed of dense subepithelial rhabdomyoblast aggregates. A-RMS cells line up along thin membranes similar to the lung alveoli. In addition, P-RMS encompasses malignant large spindle cells oriented in fascicles (Figures 13.1–13.3). FIGURE 13.1 Embryonal rhabdomyosarcoma with tight sheets of small round cells. FIGURE 13.2 Alveolar rhabdomyosarcoma with loose aggregates of small round cells. FIGURE 13.3 Pleomorphic rhabdomyosarcoma with fascicles of large spindle cells. Immunohistochemistry Muscle-specific proteins such as actin (95% of RMS), myosin, polyclonal desmin (99% of RMS), myoglobin (78% of RMS), Z-band protein, and myogenic differentiation 1 (MyoD1) can be identified by IHC.40,41 Myogenin, present in 95% of tumors, is more often expressed in A-RMS and is an independent marker of poor survival in the pediatric setting.42,43 Cytogenetics and Molecular Biology Molecular studies such as RT-PCR and FISH identify characteristic fusion proteins in ambiguous cases, especially in A-RMS where FISH for FOXO1 rearrangement became a necessary test to confirm diagnosis based on the recommendations of the Soft Tissue Sarcoma Committee of the COG. Nowadays, FISH or RT-PCR for FOXO1 rearrangement is performed on all RMS tumors. This detailed diagnostic approach is summarized in Exhibit 13.1, taken from a 2018 report from the COG Soft Tissue Sarcoma Committee.44 RMS SUBTYPES: CLINICAL, PATHOLOGIC, GENETIC, AND MOLECULAR FEATURES The International Classification of Rhabdomyosarcoma was derived from detailed pathologic study of patients who were enrolled in the first four Intergroup Rhabdomyosarcoma Study Group (IRSG) 176therapeutic trials (IRS-I to -IV).31,40 According to the 2015 WHO classification of skeletal muscle tumors in pediatric RMS, four major histologic subtypes with characteristic treatment strategies and prognostic implications were described.45 EXHIBIT 13.1 Rhabdomyosarcoma Diagnostic and Staging Workup BMA/Bx, bone marrow aspiration and biopsy; CSF, cerebrospinal fluid; FP RMS, fusion-positive RMS; LN, lymph node; MyoD, myogenic differentiation; RMS, rhabdomyosarcoma; RPLND, retroperitoneal lymph node dissection. Source: Reproduced with permission from Borinstein SC, Steppan D, Hayashi M, et al. Consensus and controversies regarding the treatment of rhabdomyosarcoma. Pediatr Blood Cancer. 2018;65(2):e26809. doi:10.1002/pbc.26809 Embryonal RMS As mentioned in the section “Incidence,” E-RMS affects young children and is mostly found in the head, neck, and GU sites. This entity accounts for 59% of all RMS cases. Eighty-four percent of E-RMS cases are of the classic subtype. The botryoid variant is diagnosed in 10% of cases. Typical rhabdomyoblasts have a moderate to deeply eosinophilic cytoplasm representing poorly formed myofilaments, and are arranged in sheets and large nests, with infrequent intermixed fusiform cells (Figure 13.1). E-RMS lacks specific translocations, but usually has loss of heterozygosity (LOH) at the 11p15 locus, the site of insulin-like growth factor 2 (IGF-2), an imprinted gene. This results in upregulation of IGF-2 expression in tumor cells, which in turn stimulates tumor growth.46–49 Other genetic and epigenetic aberrations in E-RMS include the following: • Alteration of genes at 11p15.5 locus (in addition to IGF-2): H19, CDKN1C, and HOTS • ALK copy gain • Mutations in pRb, TP53, GLI, CDKN2A, CDKN2B, RAS, FGFR4, PIK3CA, CTNNB1 (beta-catenin), and MyoD1 • Deletion in NF1 gene • DNA methylation aberrations Botryoid Variant Sarcoma botryoid has a grape-like gross appearance. It only affects infants and typically arises from the wall of the bladder or the vagina. Spindle Cell/Sclerosing RMS Spindle cell/sclerosing RMS (S-RMS) has been recently identified as a new subtype. However, patients are still stratified based on more established risk factors while awaiting new data on treatment modalities. These forms are more prevalent in males. Poor prognostic features include adult age, parameningeal tumors, and the presence of MYOD1 mutations.50,51 The spindle variant occurs in infants younger than 1 year of age. When needed, IHC for desmin, myogenin, and MyoD1 should be performed to distinguish this tumor from congenital/infantile fibrosarcoma. Unique translocations with rearrangements of VGLL2 and NCOA2 facilitate diagnosis. Infants <1 year of age with this particular histology usually have a favorable outcome. The sclerosing variant is an aggressive, poor prognostic form occurring in children older than 1 year of age as well as in adults. Mutations in MyoD1 and/or PI3KCA genes are characteristic. 177Alveolar RMS A-RMS typically affects the trunk or the extremities in children. It has a poorer prognosis than E-RMS with 5-year survival rates barely reaching 50%. Similar to pulmonary alveoli, the morphology of A-RMS consists of pseudoalveolar spaces separated by fibrovascular septa that are lined with round, loosely adherent rhabdomyoblasts. The latter frequently shed into these spaces. A-RMS is characterized by a specific translocation t(2;13)(q35; q14) between the following: • The long arm of chromosome 2 that includes the PAX3 gene, a transcription regulator during early neuromuscular development and • The long arm of chromosome 13 that harbors the FOXO1 gene, a transcription factor52,53 Several cases have been reported with a variant t(1;13)(p36;q14) translocation, resulting in a fusion between PAX7 and FOXO1 genes.54 PAX3 and PAX7 encode related DNA-binding domains; the resulting fusion transcript promotes the development of a transformed phenotype through mechanisms that are yet to be determined.55 FISH test using a FOXO1 probe and/or RT-PCR assays that detect the presence of either of these fusion genes are both rapid and accurate tests for identifying these translocations. Of 78 children with A-RMS, Sorensen et al. detected PAX3 and PAX7 fusion transcripts in 55% and 22% of cases, respectively. Twenty-three percent were fusion negative. They also demonstrated a higher risk of failure (p = .025), death (p = .019), and increased bone marrow involvement in metastatic tumors harboring a PAX3–FOXO1 translocation.56,57 Conversely, the PAX7–FOXO1 translocation occurs in younger patients, affects the extremities, and has better outcomes. According to the Soft Tissue Sarcoma Committee of the COG, the diagnosis of A-RMS requires the presence of a typical architectural pattern in >50% of the tumor as well as the presence of a FOXO1 rearrangement, t(1;13) or t(2;13), by FISH or RT-PCR (when cytogenetic testing is unavailable). The latter is also beneficial for risk stratification as well as confirmation of diagnosis in the ambiguous cases. Other genetic/epigenetic abnormalities include the following: • PAX3/FOXO4, PAX3/NCOA1, PAX3/NCOA2, and FOXO1/FGFR1 translocations • CDK4 amplification • TP53, CDKN2A, CDKN2B mutations • FGFR4 and ALK copy gains • RASSF, HIC1, CASP8 tumor suppressor mutations • DNA methylation: Compared to E-RMS, A-RMS has a distinct DNA methylation pattern and is enriched in DNA hypermethylation of polycomb target genes Up to 45% of RMS with alveolar morphology lack FOXO1 (or FKHR) rearrangement or other known genetic abnormalities and often behave like E-RMS with a more favorable outcome.58,59 This suggests that fusion gene status regardless of histology is an essential factor in risk stratification of RMS. Adult patients with FN RMS have a better prognosis compared to FP RMS. Unlike in children, no significant difference in prognosis between PAX3/FOXO1 and PAX7/FOXO1 fusions was detected. In consequence, nonpleomorphic RMS could be classified into two major subgroups: FN RMS and FP RMS. Pleomorphic RMS P-RMS is the subtype that mainly affects adults. The risk factors, biology, and genetics remain unclear because of the low incidence of this disease and the lack of corresponding studies in adults. Tumor architecture consists of large spindle cells that may resemble other types of spindle cell sarcomas (pleomorphic myogenic soft tissue sarcoma). This subtype is extremely genetically heterogeneous. Noujaim et al. published a series of 45 adult patients with P-RMS. Median age at diagnosis was 71.5 years. Median OS was 12.8 and 7.1 months in localized and metastatic disease, respectively. Response rates to pediatric chemotherapy protocols were low and relapse rate was high (53.8%).60 STAGING Two major staging systems are used in newly diagnosed pediatric RMS: Clinical Group (CG) and TNM.61 Conversely, traditional soft tissue sarcoma staging systems are still used in adults. CG Classification Developed by the IRSG in 1972, this widely used surgical pathologic staging system (Table 13.1) includes four disease categories with different prognoses.62,63 It evaluates the quality of the surgical 178resection based on the presence of residual disease and microscopic LN involvement. While it helps define treatment strategies, this classification does not consider other prognostic factors such as age, primary site, histology, cytogenetics, tumor size, and nodal involvement. TABLE 13.1 Clinical Group and Description Total gross resection with evidence of regional spread a. Grossly resected tumor with microscopic residual disease (tumor at margin) b. Regional involved nodes, completely resected with no microscopic residual disease c. Regional involved nodes and microscopic residual disease TNM Classification This classification (Table 13.2) was developed and incorporated into the fourth IRSG protocol (IRS-IV).64 Patients are assigned to one of four stages according to the site and size of the primary tumor, regional node involvement, and the absence or presence of distant metastases. Favorable disease sites include the orbit and eyelid, other nonparameningeal head and neck locations, and nonbladder, nonprostate GU tumors (e.g., paratesticular tumors), while unfavorable sites include the extremities, bladder and prostate, cranial parameningeal sites, trunk, and retroperitoneum. 179The TNM and CG staging systems complement each other, and both of them are used to assess prognosis and select treatment for individual patients with RMS. PROGNOSTIC FACTORS According to the SEER program, mortality of RMS patients is predominantly related to age, tumor site, histology, and disease extension. Age In the past decades, mortality rate from cancer in children has been cut by half. However, these favorable results are yet to be achieved in adolescents and adults. The 5-year OS rate of RMS is 67% in children, 51% in adolescents, and 27% in adults. The most favorable age group is between 1 and 9 years with a 5-year survival rate of 87% according to the IRSG trials. Infants younger than 1 year of age and children older than 9 years have a 5-year survival rate of 76%. Among the adult population, the 5-year OS is inversely proportional to age and reaches 36%, 29%, and 11% in young adult, middle-aged, and elderly patients, respectively. According to the SEER database of the National Cancer Institute (NCI), adults tend to have unfavorable histologic subtypes (pleomorphic subtype) at unfavorable sites (65% vs. 55%, p < .0001). They also have overall worse survival than children for similar subtypes, especially in the nonmetastatic setting (5-year survival rate 47% vs. 82%).65 Tumor Site Prognosis is linked to the primary tumor site, but not always to the possibility of resection. In fact, orbital and GU tumors have a better prognosis than those occurring in the extremities, retroperitoneum, or trunk. Histology Classically, E-RMS is correlated with a better prognosis than either A-RMS or P-RMS. A-RMS is more often diagnosed in unfavorable age groups or locations (<1 year of age or older patients, extremities, metastatic sites). Adult P-RMS is a particularly aggressive undifferentiated tumor, with a behavior similar to undifferentiated pleomorphic soft tissue sarcoma. It is treated suboptimally with pediatric protocols, hence the dismal prognosis (Figure 13.4). Molecular Biology PAX3/PAX7–FOXO1 fusion is observed in A-RMS. FN A-RMS tends to have a better outcome, more consistent with E-RMS, and is often reclassified as such. FP tumors involving PAX7 may have a better outcome than those with PAX3 fusions (Figure 13.4). Disease Extension Patients presenting with metastatic disease have a worse prognosis than those with localized disease. Moreover, patients with metastatic disease lacking other poor risk factors known as the Oberlin criteria, such as unfavorable sites, more than three sites, bone marrow involvement, and age younger than 1 year or older than 10 years, have a higher event-free survival (EFS) than those with three to four adverse features (50% vs. 10%).66 Prognostic Models: the IRSG Model Risk-based algorithms help select treatment in pediatric RMS. There are currently no risk-based models for adults. The IRSG, now known as the Soft Tissue Sarcoma Committee of the COG, developed a recurrence risk-based model to refine therapy in children. Favorable prognostic factors include absence of metastases; orbital, nonparameningeal head/neck, and GU nonbladder/non-prostate primary sites; grossly complete surgical removal of localized tumor at the time of diagnosis; embryonal/botryoid histology; tumor size ≤5 cm; and age younger than 10 years at diagnosis67 (Table 13.3). Other classifications exist such as the one used in European RMS studies that define subgroups from A to H.68 More recent COG protocols (ARST0431 and ARST1431) have a distinct risk-based classification with intermediate- and high-risk groups that were adapted to each protocol. FIGURE 13.4 Prognosis of rhabdomyosarcoma according to histology and molecular biology. A-RMS, alveolar RMS; E-RMS, embryonal RMS; RMS, rhabdomyosarcoma. TREATMENT OF PEDIATRIC AND ADULT RMS Common Treatment Strategies in Children and Adults All patients diagnosed with RMS should be treated in experienced centers. The treatment strategy (type of treatment, timing, intensity) should be elaborated by a multidisciplinary sarcoma board that includes an experienced pathologist, a surgeon, a radiation oncologist, and a medical oncologist. Decisions should also be discussed directly with the patient. They often involve a multimodal approach that combines surgery, chemotherapy, and RT. Short- and long-term toxicities should be taken into consideration. All patients should be encouraged to participate in clinical trials. Pediatric RMS Micrometastatic disease is often present at diagnosis of RMS. The addition of chemotherapy and RT to surgery increased cures rates in children from 20% to 70%. Nowadays, treatment of pediatric RMS is based on chemotherapy in the neoadjuvant and adjuvant settings, surgery if possible, and RT to improve local control. Large cooperative international groups such as the COG in the United States and the more recently founded European Pediatric Soft Tissue Sarcoma Study Group (EpSSG) in Europe developed several multimodality treatment protocols based on the estimated risk of disease recurrence. Risk-adapted therapy uses prognostic tables such as the IRSG risk classification table (Table 13.3) to decide treatment strategy.2,62,69 Surgery Patient outcomes depend on the extent of surgical resection, which should be complete in localized disease if functional and cosmetic results are acceptable. If a resection with negative margins is not possible or if RMS involves the orbit, biliary tract, or the GU tract (except for paratesticular RMS), neoadjuvant chemotherapy should be administered. Clinically and radiographically suspicious LNs should be excised. Given the low risk of LN involvement, routine cervical sampling is not mandatory in head and neck tumors (except orbital tumors). On the other hand, up to 47% of adolescents with radiographically negative retroperitoneal LNs have pathologic documentation of LN metastasis. Therefore, all boys after the age of 10 with paratesticular 182RMS should undergo ipsilateral nerve-sparing retroperitoneal LN dissection along with radical inguinal orchiectomy. Moreover, patients with positive nodes usually receive additional RT and cyclophosphamide, while those with negative nodes are only given a two-drug regimen (vincristine dactinomycin [VA]).33,70 Sentinel LN biopsy is recommended in patients with extremity RMS.71 Re-excision should be considered in case of positive resection margins. Second-look surgery to document the histopathologic response to chemotherapy did not demonstrate any survival benefit. However, it may be considered in children with CG III disease to identify those who may benefit from reduced-dose RT.72 Survival advantage from resection of distant metastases has not been proved yet. The decision to perform metastasectomy should be thoroughly discussed at the tumor board on a case-by-case basis. Radiation Therapy RT is recommended for local control in all patients with RMS, except those who have embryonal/FN (any site) CG I disease. Three-dimensional (3D) conformal RT along with intensity-modulated RT should be privileged to minimize toxicities. Proton beam RT significantly reduces late toxicities in orbit and parameningeal tumors.73 Tumor volume should include at least a 1-cm margin of normal tissue as well as involved LNs. Although no studies have shown a benefit for specific timing, radiation is usually initiated after four cycles of chemotherapy, even in patients with cranial nerve palsy or skull base erosion from parameningeal RMS.74 Radiation doses are adapted according to the CG and the LN involvement. Several attempts to decrease RT doses to limit long-term toxicities in various tumor locations have been made, some with encouraging results. Craniospinal RT is indicated when tumor cells are found in the cerebrospinal fluid. Whole-brain RT should be delivered in case of several brain metastases and a negative CSF cytology. Brachytherapy should be considered for small critically located tumors (GU, head and neck).75,76 In addition, RT may be used in metastatic disease to control both the primary and metastatic sites. Some protocols even recommend whole-lung RT (generally up to 14.4 Gy) for patients with multiple lung lesions. Chemotherapy All patients with RMS should receive chemotherapy along with local treatment. Chemotherapy may be administered both in the neoadjuvant setting (induction treatment) to facilitate subsequent surgery and to test for tumor chemosensitivity. It can also be given in the postoperative setting to eradicate micrometastases and prevent recurrence. A classic chemotherapy regimen in pediatric RMS includes an alkylating agent (cyclophosphamide or ifosfamide), vincristine, and dactinomycin. None of the trials performed since the 1970s has been able to identify a chemotherapy protocol more effective than vincristine, dactinomycin, and cyclophosphamide (VAC) or ifosfamide, vincristine, and dactinomycin (IVA). Based on the COG studies, the VAC regimen is considered standard of care in North America. Intensification of VAC by adding other agents such as etoposide, ifosfamide, topotecan, melphalan, and doxorubicin failed to improve outcomes compared with VAC.77,78 The ARST0531 trial compared the alternation of VAC and VI (vincristine and irinotecan) to standard VAC in patients with intermediate-risk disease (patients with nonmetastatic alveolar tumors and patients with stage 2 or 3, group III embryonal tumors). OS and EFS were similar between the two arms; however, the VAC/VI arm was associated with less hematologic toxicity and lower cumulative cyclophosphamide doses.79 On the other hand, the EpSSG replaces cyclophosphamide with ifosfamide because of the lower gonadal toxicity rates with the former. IVA is the European standard of care in pediatric RMS. The EpSSG RMS 2005 trial is the first multicenter, randomized controlled, open-label Phase 3 study that investigated the benefit of adding doxorubicin 30 mg/m2 given as a 4-hour infusion on days 1 and 2 to a standard IVA regimen, as well as the benefit of a maintenance treatment in patients with high-risk localized disease. Unfortunately, no survival benefit was found and toxicity was worse in the IVA plus doxorubicin group (IVAdo). Table 13.4 features the most commonly used regimens for pediatric RMS. Treatment strategies according to risk group (North American experience) are discussed in the following sections (Exhibit 13.2). In all the risk groups, RT, when indicated, is usually started at week 13 of chemotherapy. Dose reductions are necessary for children younger than 3 years of age. Dactinomycin administration should be held during RT and resumed thereafter. Low Risk Subset A This category includes embryonal and alveolar FN, stage 1/2, CG I/II tumors, as well as stage 1, CG III orbital tumors. Three-year failure-free survival (FFS) rate exceeds 85%.80 Fifteen to sixteen cycles of VA (as per regimen D9602)81 or four short cycles of VAC followed by four cycles of VA (as per regimen ARST0331)82 could be administered. 183TABLE 13.4 Most Frequently Used Chemotherapy Regimens in Pediatric Rhabdomyosarcoma
Clinical Group I
Localized disease, completely resected, regional nodes not involved. This includes both gross inspection and microscopic confirmation of complete resection
Clinical Group II
Clinical Group III
Incomplete resection or biopsy with gross residual disease
Clinical Group IV
Distant metastatic disease present at onset

Related posts:
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Rhabdomyosarcoma
Pamela Abdayem and Sarah Dumont