A malignant tumor of mesenchymal cell origin is called a
sarcoma. Mesenchymal cells normally mature into skeletal muscle, smooth muscle, fat, fibrous tissue, bone, and cartilage.
Rhabdomyosarcoma (RMS), represented as two major histological subtypes with “alveolar” and “embryonal” morphological features, is thought to arise from immature mesenchymal cells that are committed to skeletal muscle lineage, but these tumors can also arise in tissues in which striated muscle is not normally found, such as in the urinary bladder.
Undifferentiated sarcomas are mesenchymally derived tumors that cannot be ascribed to any specific lineage. Some tumors may also display multilineage markers; examples include ectomesenchymomas, which are tumors with evidence of both skeletal muscle and neuronal lineage,
1 and malignant triton tumors, which are malignant peripheral nerve sheath tumors (schwannomas) with evidence of rhabdomyoblastic elements. These tumors are discussed in greater detail elsewhere.
The incidence of RMS is slightly less than half that of all other forms of non-RMS softtissue sarcomas (NRSTSs) combined. Important epidemiologic, biologic, and treatment differences exist both within the family of RMS and between RMS and NRSTS. Modern cooperative group studies use a modified (site-based) tumor-nodes-metastasis (TNM) staging system, comparable to what has been used for adult NRSTS, in addition to a surgicopathologic staging system (Clinical Group [CG]), to define groups of patients at low-, intermediate-, and high risk of treatment failure. The development of large-scale international collaborative multimodality therapeutic protocols for treating these tumors was instrumental in leading to dramatic improvements in the curability of these neoplasms, especially for patients with locally extensive but unresectable tumors. Over the past two-plus decades, however, outcome has plateaued for the majority of patients with locoregional disease, and improvements in outcome for patients with metastatic or recurrent tumors have proven elusive. As treatments have become more effective at prolonging survival for nearly all patients, and producing cures in the majority, there has been an increased awareness of, and focus on reducing, the short- and long-term sequelae of therapy.
EPIDEMIOLOGY AND GENETIC SUSCEPTIBILITY
The annual incidence of RMS in children 19 years of age or younger is 4.9 cases per million children, with approximately 400 new cases diagnosed in the United States each year.
2 Among the extracranial solid tumors of childhood, RMS is the third most common neoplasm after neuroblastoma and Wilms tumor. Almost two-thirds of cases of RMS are diagnosed in children 9 years of age or younger, with a smaller incidence peak in early mid-adolescence. In all age groups, the tumor is slightly more common in males than in females.
2 A 2009 analysis compared 2,600 cases of adult (20 years of age and older) and pediatric RMS captured over 22 years in the Surveillance, Epidemiology and End Results (SEER) Program.
3 The incidence of RMS in adults was substantially lower than in children, and approximately 40% of the total number of cases were in adults, suggesting that there are slightly more than 550 new cases of RMS diagnosed each year in the United States in patients of all ages. A higher proportion of adults had one or more “unfavorable” prognostic features; although potentially important differences in treatment could not be ruled out as a contributing factor, pediatric patients had a consistently better outcome than adult patients. A more recent analysis of outcome in adult RMS patients treated at a single institution suggested that survival was significantly improved, and comparable to that of pediatric patients, when adults were treated on or as per an RMS therapeutic protocol.
4 A separate analysis of SEER data between 1975 and 2005 for nearly 1,000 children and adolescents 19 years of age and younger revealed a stable incidence of embryonal RMS (ERMS) over this time period, but a significant increase in the incidence of alveolar RMS (ARMS), possibly the result of shifting diagnostic criteria.
5 This analysis confirmed a bimodal age peak for ERMS, though the second peak during adolescence was noted for males only; in this analysis, ARMS incidence did not vary by age or gender. Improved survival over this period was seen only for cases of ERMS.
An international study confirmed previous reports of racial and gender differences in the incidence of RMS.
6 In the United States, the incidence of RMS for African-American females was found to be only half that for Caucasian females, whereas the rate for males was similar in both ethnic groups. The incidence of RMS in most of Asia appears to be lower than among mainly white populations in Western industrialized countries.
Although these tumors may arise virtually anywhere in the body, there are certain distinctive clusters of features regarding age at diagnosis, site of primary tumor, and histology. For example, head and neck tumors are most common in children younger than 8 years of age and, if arising in the orbit, are almost always of the embryonal variety. On the other hand, extremity tumors are more commonly seen in adolescents and are more frequently of the alveolar subtype. A unique form of RMS arising from the bladder or vagina, the botryoid variant (so named because of its resemblance to a protruding cluster of grapes), is seen almost exclusively in younger children.
Until recently, published studies of potential etiologic factors related to the development of RMS consisted of relatively small series. Although the overwhelming majority of cases of RMS occur sporadically, the development of RMS has been associated with certain familial syndromes, such as neurofibromatosis and the Li-Fraumeni syndrome (LFS), which includes familial clustering of RMS and other soft tissue tumors in children, with adrenocortical carcinoma and early-onset breast carcinoma in adult relatives.
7 The LFS has been associated with germline mutations of the
p53 tumor suppressor gene.
8 In a study of 33 cases of sporadic RMS, 3 of 13 children younger than 3 years of age at diagnosis (compared with none of the 20 children older than 3 years of age) were found to have germline mutations in their
p53 gene.
9The overall risk of a genetic predisposition to cancer has been estimated to be 7% to 33%, based on the patterns of cancer in the families of 151 children with soft tissue sarcomas.
10 These included syndromes that appeared to be independent of p53 abnormalities. Of further interest, RMS has been observed in association with Beckwith-Wiedemann syndrome, a fetal overgrowth syndrome associated with abnormalities on 11p15, where the insulin-like
growth factor II (
IGFII) gene is located.
11 (Also see
Chapter 2.) Some of the factors that may play a role in these phenomena are discussed later in this chapter.
Studies of children with Costello syndrome, a rare congenital disorder characterized by postnatal growth retardation, typical coarse facies, loose skin, and developmental delay, have noted an increased risk for development of solid tumors, most commonly RMS. Costello syndrome is caused by heterozygous de novo point mutations in
HRAS, resulting in increased activation of the mitogen-activated protein kinase pathway.
12,13 Of about 100 known Costello syndrome patients, 10 RMSs have been reported.
14,15 A review of IRS (Intergroup RMS Study) cases reported 5 cases of RMS patients with NF-1 out of the 1,025 cases enrolled in IRS-IV.
16 Results of a large national case-control study were reported in 1993; this study involved 322 RMS patients younger than 20 years of age who were enrolled in IRS-III and an equal number of randomly selected age-, sex-, and race-matched controls.
17 Use of marijuana by the mother in the year before the child’s birth was associated with a threefold increased risk of RMS in the child, and maternal cocaine use was associated with a fivefold increased risk. Use of marijuana, cocaine, or any recreational drug by the father was also associated with an approximately twofold increased risk. Consistent with a potential interaction between genetic susceptibility (e.g., germline p53 mutations) and environmental factors in the development of some cases of RMS, use of cocaine by the mother and use of marijuana by both parents were associated with a significantly earlier age at diagnosis of RMS (compared with all children in IRS-III). A more recent case-control study conducted by the Children’s Oncology Group (COG) involving more than 300 RMS cases and an equal number of matched controls confirmed an association between prenatal X-ray exposure and the subsequent development of RMS, with the strongest association seen between first-trimester exposure and the subsequent development of ERMS (relative risk, 10.5; 95% confidence interval [CI], 1.5 to 458.4).
18
MOLECULAR BIOLOGY
It appears likely that multiple molecular genetic alterations involving both muscle differentiation pathways as well as cell proliferation pathways lead to the development of RMS. As the details of these pathways become better defined, it is becoming increasingly clear that alterations in the FGFR4/RAS/PIK3CA pathway are commonly altered and may represent a common “targetable” pathway for these tumors.
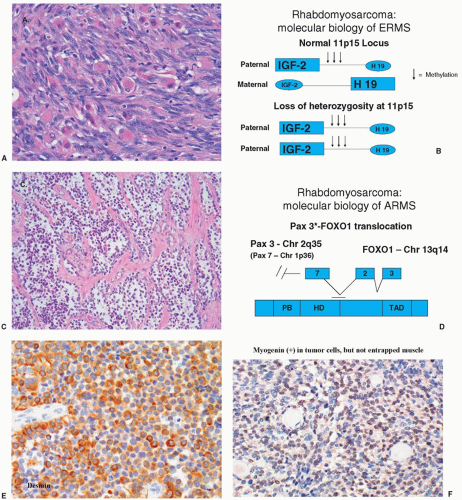
19,20The two major histologic subtypes of RMS, namely, ERMS and ARMS, have been found to have distinct genetic alterations that are presumed to play a role in the pathogenesis of these tumors. ARMS has been demonstrated to have a characteristic translocation between the long arm of chromosome 2 and the long arm of chromosome 13, referred to in shorthand notation as t(2;13)(q35;q14).
21 This translocation has been molecularly cloned and shown to involve the juxtaposition of the
PAX3 gene on chromosome 2q35 (or, rarely, the
PAX7 gene located at chromosome 1p36 leading to a t(1;13)(p36;q14)), believed to regulate transcription during early neuromuscular development, and the FKHR gene, also known as FOXO1A, a member of the forkhead family of transcription factors.
22,23 It is presumed that the consequence of this fusion transcription factor PAX3/7-FOX1A (PF) is the abnormal activation of genes that contribute to the transformed phenotype. Recent application of molecular techniques has now clearly demonstrated that the presence or absence of the PF fusion demarcates two distinct molecular forms of RMS.
PAX3/7-FOXO1 fusion-positive RMS (PFP) tumors make up the ARMS subset (see section on pathology) and clearly have a lower burden of mutations than do PAX3/7-FOXO1 fusion-negative (PFN) tumors.
20,24 The PFP tumors commonly have overexpression/amplification of MYCN as well as amplification of the cyclin-dependent kinase CDK4, and this combination of alterations appears to be a common underlying feature of these tumors.
25,26 The PF fusion transcription factor has also been found to upregulate a number of genes that are altered in PFN ERMS tumors including c-MET, MYOD1, and FGFR4 as well as the IGFIR.
20,27,28 This finding is of particular interest since it is suggestive of a final common pathway for both PFP and PFN RMS.
PFN ERMS is known to have loss of heterozygosity (LOH) at the 11p15 locus.
29 It has been shown that this LOH involves loss of maternal genetic information with duplication of paternal genetic material at this locus. This region is of particular interest because it is the location of the
IGF-II gene, which codes for a growth factor believed to play a role in the pathogenesis of RMS (see later discussion).
IGF-II has been demonstrated to be imprinted with only the paternal allele being transcriptionally active.
30,31 It is therefore conceivable that in this tumor, LOH with paternal disomy may lead to overexpression of
IGF-II. However, it is also possible that LOH at 11p15 may reflect the loss of a tumor suppressor activity that has not been identified, or that both activation of
IGF-II and loss of tumor suppressor activity result from LOH at 11p15 in ERMS.
31The most frequently observed oncogene abnormalities seen in RMS are RAS mutations. Activated forms of both NRAS and Kirsten-RAS (KRAS) have been isolated from both RMS cell lines as well as from tumor specimens.
32 A survey of ERMS tumor specimens found a 35% incidence of either activated NRAS or KRAS.
33 Of particular note, recent data suggest that RAS mutations in ERMS are associated with high-risk ERMS and that low-risk tumors rarely if ever harbor RAS mutations.
24 The finding of RAS activation has recently been confirmed by investigators who created an RAS-induced zebrafish model of RMS (see Animal Models section) and noted an “RAS induced gene signature” that was evident in human ERMS as well.
34Other recurrent mutations found in PFN ERMS include FGFR4, PIC3CA, NF1, TP53, CTNB1, and BCOR.
19,20,35The discovery of the myogenic basic helix-loop-helix (bHLH) MyoD family of proteins (including MyoD, myogenin, Myf5, and MRF4) has greatly enhanced understanding of normal skeletal muscle differentiation.
36 These proteins function to commit mesenchymal cells to a skeletal muscle lineage and to activate their terminal differentiation program by inducing transcription of skeletal muscle-specific proteins such as myosin and creatine kinase. The almost universal expression of MyoD family proteins in RMS provides further strong evidence of the skeletal muscle lineage of these tumors and has allowed for further refinement in the classification of pediatric sarcomas. For example, some cases that would previously have been called undifferentiated sarcomas can be classified as RMS based on expression of these lineage-specific transcription factors.
The failure of RMS cells to terminally differentiate and growth arrest despite the expression of these bHLH proteins has raised the question of what is altered in this pathway compared with normal skeletal muscle cells. A recent intriguing study found that 10% of ERMS tumors sequenced were noted to have a Leu122Arg mutation in MYOD1 that functions to block wild-type MYOD1 activity and may in part explain a differentiation block in this subset of tumors.
28
PATHOLOGY
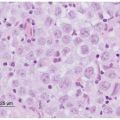
RMS represents one of the small round blue cell tumors of childhood, among which the pathologist typically discerns individual subtypes using morphological features assessed by light microscopy and aided by immunohistochemical staining, molecular genetics, and, sometimes, electron microscopy. The feature defining RMS is evidence for skeletal muscle lineage specification and/or differentiation, which often consists of light microscopic identification of cross-striations characteristic of skeletal muscle, or characteristic rhabdomyoblasts. Immunohistochemical staining aids in the detection of skeletal muscle-specific proteins to discriminate RMS from other small round blue cell tumors.
42 These proteins include muscle-specific actin and myosin, desmin, myoglobin, Z-band protein, and MyoD.
43 Myogenin expression, demonstrated to be present in the majority of RMS,
44 can also be used, but its selective expression in ARMS provides added value for discriminating RMS subtypes.
45,46 Detection of the PAX3-FOXO1 or PAX7-FOXO1 rearrangement by fluorescence in situ hybridization (FISH) in tissue specimens or by reverse transcriptase polymerase chain reaction (RT-PCR) in tumor-derived RNA can clinch a diagnosis of RMS because this particular molecular genetic abnormality is specific for the ARMS subtype. It is important to emphasize that evidence of skeletal muscle gene expression is not sufficient to make a diagnosis of RMS.
47 Other childhood neoplasms like Wilms tumor and pleuropulmonary blastoma can contain rhabdomyosarcomatous elements, and myogenic genes can be expressed in malignant triton tumor and dedifferentiated chondrosarcoma and liposarcoma.
The WHO recognizes three histological variants of RMS: alveolar, embryonal, and pleomorphic, which is very similar to that originally outlined by Horn and Enterline (embryonal, botryoid [a subtype of embryonal], alveolar, and pleomorphic).
48 The two major variants in children, ERMS and ARMS, have relatively characteristic histologic appearances and specific and distinctive molecular genetic abnormalities, as described above (
Fig. 31.1). Histologic appearance, which is based on the identification of typical morphological and architectural features, usually permits straightforward discrimination (see more below). An international pathology study, carried out to assess agreement among pathologists specializing in RMS classification, highlighted the proliferation of subtle differences in diagnostic criteria that had developed since the publication of the Horn and Enterline schema,
49,50 prompting the development of the International Classification of Rhabdomyosarcoma schema.
51 The two broad RMS subtypes in children (ARMS and ERMS) were divided such that botryoid and spindle cell RMSs were recognized as ERMS variants generally associated with a superior prognosis, while ERMS and ARMS (including the solid alveolar variant) were recognized to generally be associated with intermediate and poor prognoses, respectively. Undifferentiated sarcoma, also recognized in this scheme, is no longer included with RMS by the Children’s Oncology Group.
Under the International Classification system,
51 ERMS, representing approximately two-thirds of RMS cases, is diagnosed based on a stroma-rich, less dense, spindle cell appearance, and absence of an alveolar pattern. However, a “dense” embryonal variant was recently highlighted based on its easy confusion with ARMS.
52 The
botryoid variant tends to arise from a hollow viscus, such as the bladder or vagina in infants and young children or from the nasopharynx in older children. Microscopically, it presents as a polypoid mass growing under an epithelial surface and has as its characteristic feature the presence of a dense tumor cell layer under the epithelium (the cambium layer). The
spindle cell variant tends to arise as a paratesticular tumor but may also be seen in the head and neck, extremity, and orbit.
53 The cells have an elongated, spindle appearance and grow either in a storiform pattern with abundant collagen between the tumor cells or in bundles with a low-to-moderate amount of collagen. Although botryoid and spindle cell variants are often associated with less aggressive tumor behavior than the classic embryonal disease, neither feature is incorporated into standard risk stratification schemes.
54The alveolar subtype of RMS is composed of densely packed, small, round cells lining septations that appear histologically reminiscent of pulmonary alveoli. A variant form, known as solid ARMS, has been identified in tumors that do not have alveolar septations but have cells that are small, round, and densely packed.
50 The clinical behavior of the solid alveolar variant appears to be identical to that of the conventional alveolar subtype. The degree of alveolar features needed for the diagnosis of ARMS has evolved over time. The aforementioned International Classification System allowed ARMS diagnosis when only a fraction of the tumor contained “alveolar” features. However, the relative frequency of ARMS versus ERMS increased from 30% to 41% during the D9803 trial for children with intermediate-risk disease.
55 Coupled with an increasing rate of ARMS specimens lacking the expected PAX-FOXO1 fusion transcript (see more below), Children’s Oncology Group pathologists rereviewed approximately 250 specimens originally classified as ARMS in the D9803 clinical trial for children with intermediate-risk disease, and they found that 33% were felt to actually be ERMS based on a current definition calling for at least 50% of the specimen to contain alveolar elements.
52 As discussed below, this has ramifications for the frequency of so-called fusion-negative ARMS.
Other histological features of RMS merit brief discussion. First, pleomorphic RMS is only rarely diagnosed today in children, but is a relatively common form in adults, in whom it carries a poor prognosis.
56 Some (˜13% in certain series) RMS cases have also been described to have anaplastic features, present either focally or diffusely in the tumor.
57 It was equally common among cases of ERMS and ARMS, and it predicted inferior outcome, on univariate but not multivariate analysis, for patients with intermediate-risk ERMS. A small series of 11 RMS specimens from children with germline TP53 mutation had anaplastic features, but this provocative correlation must be confirmed in a larger series.
58 Sclerosing RMS, described recently in adult and pediatric patients, is also a rare histologic subtype that may be associated with more aggressive clinical behavior and poor prognosis.
59,60,61,62,63 To date, fewer than two dozen pediatric cases have been described; a whole-genome
analysis of a left deltoid tumor from a 7-year-old boy revealed an aneuploid profile with amplification of the
MDM2-HMGA2 locus at 12q13 to 15.
64 It remains unclear whether sclerosing RMS is a variant of ERMS or ARMS, or an entity unto itself.
65,66Although much past effort has focused on the importance of histological diagnosis, growing insight into RMS biology points toward molecular subtypes as crucial determinants of clinical behavior in RMS. Indeed, several recent reports demonstrate that the molecular genotype—whether the RMS specimen carries or lacks the characteristic PAX3-FOXO1 or PAX7-FOXO1 fusion transcripts—correlates more closely with molecular and clinical phenotype than histology. For example, when broadly evaluating the “signature” of expressed genes in RMS, so-called fusion-negative ARMS clusters more closely to the ERMS cases than to the “fusion-positive” cases.
67 Further, in a large retrospective series
67 and in an analysis of a single clinical trial of approximately 600 children with intermediate-risk RMS (COG D9803 study), survival in children with fusion-negative ARMS matched that in those with ERMS, which exceeded survival in those children with fusion-positive ARMS.
68 Specifically, 5-year event-free survival (EFS) is worse for those with fusion-positive RMS (54% and 65% for those with PAX3- and PAX7-FOXO1, respectively) than for those with ERMS (77%) or fusion-negative ARMS (90%).
The prognostic significance posed by a specific fusion subtype (i.e., PAX3- vs. PAX7-FOXO1) in RMS remains unproven, with numerous studies yielding seemingly conflicting results. For example, two relatively small studies of “convenience cohorts” came to opposite conclusions: In one study, risk of treatment failure and death was significantly greater in children with metastatic PAX3-FOXO1 versus PAX7-FOXO1 disease,
69 while a second study showed no difference in outcome among those with tumors harboring the PAX7-FOXO1 rearrangement as compared to PAX3-FOXO1.
70 A follow-up analysis from the first group did not reveal a survival advantage associated with fusion subtype; indeed, this group showed that survival was better for those patients with tumor tissue
merely available for analysis,71 which highlights the problem with studies of so-called convenience cohorts.
72 In an attempt to surmount this obstacle, the aforementioned COG D9803 study reported survival in children with intermediate-risk RMS that had either PAX7- or PAX3-FOXO1 disease. As noted earlier, the 5-year EFS tended to be better for those with PAX7-FOXO1-positive RMS, but such a conclusion is tenuous given the small number of patients with PAX7-FOXO1 disease. It should be emphasized that, even though the significance of specific fusion gene is not clear, most experts now feel that the presence or absence of any fusion gene should be incorporated into risk stratification in RMS clinical trials.
54,68How exactly is the diagnosis of RMS made? The historical standards hold that diagnosis of RMS requires tumor tissue that, as noted above, is scrutinized by light microscopy with a battery of histological and immunohistochemical stains to arrive at a diagnosis. Such analysis requires biopsy by incisional/excisional procedures or core needle sampling. Although this remains the standard, new insights into disease biology and new tools are beginning to challenge this standard approach. For example, it has long been recognized that the presence of a PAX-FOXO1 rearrangement could be used to confirm a diagnosis of ARMS. Such translocations are now readily detected using commercial FISH probes and routine pathology material. As an alternative, RNA extracted from frozen or formalin-fixed, paraffin-embedded tumor can be used to detect a fusion transcript by RT-PCR techniques. In a series of approximately 125 COG patients with ARMS, a chromosomal translocation or fusion transcript was detected in 83% of the cases,
68 excluding the occasional cases in which either FISH or RT-PCR failed due to technical reasons, and the results were completely concordant between two technical approaches. Given the biological importance of fusion status over histology, one might argue that detection of the fusion might supplant the need for morphological assessment, and this could be accomplished with much less tissue. Pushed to an extreme, this concept suggests that fine-needle aspiration might be utilized, especially if coupled to immunohistochemical staining and/or molecular diagnostic studies. This is supported by a number of reports of single cases and small series,
73 but has not risen to wide, routine practice.
Regardless of the type of tissue obtained, how fusion status is best determined is not totally clear because the focused approaches noted above will miss the rarer, but potentially important, “variant” translocations, such as those involving NCOA1 and NCOA2.
74,75 Although one obvious solution is to expand the portfolio of clinically validated FISH probes or RT-PCR assays to detect these variants, an alternative approach would be to develop a surrogate for fusion gene testing that might capture the biology imposed by the fusion protein. RNA gene expression profiling shows that “signatures” of expressed transcripts correlate with fusion status,
76,67 and one report indicates that as few as two or three mRNA species, detected by a highly quantitative, digital detection of color-coded RNA probes, can discriminate between fusion-positive and fusion-negative RMS using clinical-grade formalin-fixed, paraffin-embedded specimens.
77 As a potentially simpler—though perhaps not as quantitative—approach, several reports have shown that immunohistochemical detection of a small panel of proteins also has the capacity to discriminate fusion-positive and fusion-negative RMS. For example, one study exploring the diagnostic and prognostic utility of a gene expression-based immunohistochemistry panel for testing 252 RMS tumors and 2 markers (AP2β and p-Cadherin for ARMS, and epidermal growth factor receptor and fibrillin-2 for ERMS) proved both highly sensitive and specific for subgroup determination, and appeared to be useful for stratifying patients into prognostically distinct groups.
46 A second group developed an algorithm in which quantifying myogenin, AP2, HMGA2, and NOS-1 predicted RMS fusion status with 99% accuracy.
45 The usefulness of such surrogates still needs to be prospectively validated, though.
Developing robust tools for fusion status testing is increasingly recognized to be important for RMS risk stratification, but this applies largely to ARMS histology tumors. One recent report, though, demonstrated that the expression of a “metagene” composed of just five transcripts (MG5) correlates with overall survival in children with fusion-negative RMS (hazard ratio [HR], 7.9; 95% CI, 1.9 to 33.0).
78 If computational tools can be derived such that the metagene can be developed into a score that can be applied to an individual patient, RNA-based detection of these analytes could be incorporated with other clinical features to refine risk stratification for fusion-negative RMS.
PATTERNS OF SPREAD AND CLINICAL PRESENTATION
With the completion of six generations of US cooperative group clinical trials over the past four-plus decades, the biologic behavior of RMS is now well understood. In the early, prechemotherapy era, RMS had a uniformly high rate of eventual metastasis after local control measures alone,
79 with almost all patients with alveolar histology tumors eventually dying of disease.
80 In the current era of combined modality therapy, the development of effective adjuvant chemotherapy regimens has led to a clearer picture of the patterns of treatment failure.
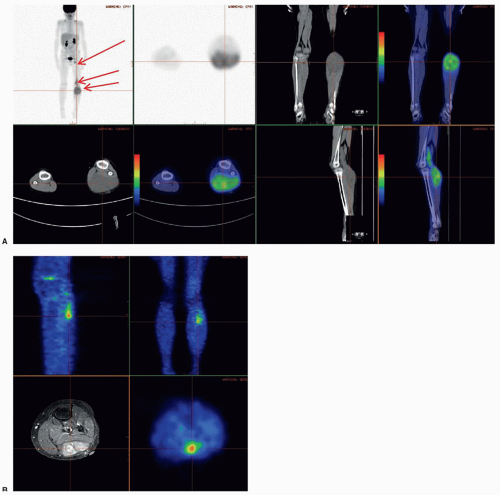
81 Between 15% and 25% of newly diagnosed patients have distant metastases, and almost half of those patients have only a single site of involvement (most commonly consisting of one or more pulmonary metastases). The lung is the most frequent site of metastasis (40% to 50%); less common sites, either isolated or in conjunction with multimetastatic disease, are bone marrow (20% to 30%), bone (10% to 15%), and, depending on the site of the primary tumor, lymph node (up to 40%).
82,83,84 Visceral organ metastases are rare in newly diagnosed patients. Factors associated with an ultra-low risk of distant spread at diagnosis
include embryonal histology and tumors without local invasion (T1), whereas alveolar histology with local invasion (T2) is associated with an increased risk of distant metastases. The presence of regional nodal metastases predicts a substantially higher risk of distant metastases, and the presence of lung metastases increases the risk of having bone or bone marrow involvement (41% with, 6% without). An international study of 788 patients with newly diagnosed metastatic RMS defined groups of “high-risk” patients with a fourfold difference in prognosis based upon age, site, histologic subtype, regional nodal spread, and the number of sites of distant metastases.
85 Although uncommon at diagnosis, visceral metastases (e.g., brain, liver) may be seen in up to 25% of terminal patients. RMS produces clinically evident signs and symptoms in two main ways: the appearance of a mass lesion in a body region without the history of temporally associated trauma and the disturbance of a normal body function by an otherwise unsuspected, critically located enlarging tumor (or enlarging regional or distant lymph nodes).
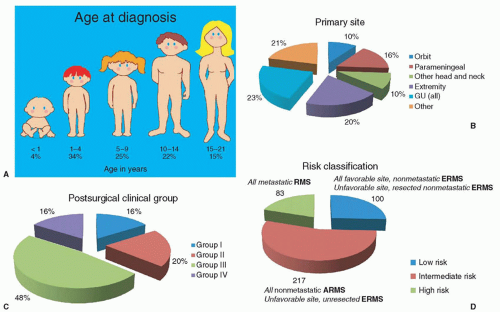
83 Typical signs, symptoms, and patterns of spread are discussed in terms of the primary tumor and are summarized here. In the first five IRS trials for which final reports are available, approximately 35% to 40% of all tumors arose from a site in the head or neck region (orbit, parameningeal [PM], other head and neck), slightly less than 25% from the genitourinary tract (bladder and prostate, vagina and uterus, paratesticular), approximately 20% from an extremity, and the remainder from truncal primaries and other miscellaneous sites (˜10% each) (
Fig. 31.2).
86,87,88,89,55
Head and Neck Region
Of the nearly 40% of RMS tumors that arise in head and neck structures, approximately one-quarter arise in the orbit or conjunctivae; 50% arise in PM sites (often referred to as “skull base” and including the nasal cavity and paranasal sinuses, pterygopalatine/infratemporal fossa, nasopharynx, and middle ear); and 25% in nonorbital, non-PM locations, such as the scalp, face, buccal mucosa, oropharynx, larynx, and neck (
Fig. 31.3). The sex ratio is almost equal, and the median age at diagnosis is approximately 6 years. At times, it can be difficult to distinguish between orbit/eyelid tumors and PM tumors since tumors arising in both locations can produce exophthalmos and occasionally ophthalmoplegia; nasal, aural, or sinus “congestion,” and/or obstruction, with or without mucopurulent or sometimes sanguinous discharge are seen primarily with PM tumors as are cranial nerve palsies, sometimes multiple, as a result of direct extension of tumor through the skull base toward the meninges.
90,91 Headache, vomiting, and systemic hypertension may result from intracranial growth of tumor after erosion of contiguous bone at the cranial base.
92 Autopsy studies show diffuse involvement of the cranial and spinal meninges reminiscent of central nervous system leukemia.
91 These tumors can also spread distantly, primarily to lungs or bones.
93 Although relatively more common for PM primary tumors, regional nodal spread is uncommonly seen in orbital and non-PM head and neck sarcomas where tumors
typically present as painless, progressively enlarging growths and tend to remain localized.
94,95,96,97
Genitourinary Tract
Genitourinary tract sarcomas are most frequently seen in the bladder and prostate (
Fig. 31.4). Bladder tumors tend to grow intraluminally, in or near the trigone, and have a polypoid appearance on gross or endoscopic examination. Hematuria, urinary obstruction, and occasionally the extrusion of mucosanguineous tissue can occur, particularly if the tumor is botryoid. Affected children are usually younger than 4 years. Prostate tumors usually produce large pelvic masses with or without urethral strangury; constipation may occur. These tumors can occur in infants or older children; even adults may be affected. Bladder tumors tend to remain localized, but prostate tumors often disseminate early to lungs and sometimes to bone marrow or bone.
Male and female genital tracts can harbor sarcoma. Vaginal tumors are commonly botryoid and are almost exclusively found in very young children who may have a mucosanguineous discharge reminiscent of that seen with a foreign body. Cervical and uterine sarcomas are diagnosed more commonly in older girls than in infants and present with a mass, with or without vaginal discharge. Regional nodal involvement (N1) is uncommon.
83 Paratesticular tumors usually produce painless, unilateral scrotal or inguinal enlargement in prepubertal or postpubertal males. The risk of tumor dissemination to regional retroperitoneal lymph nodes appears to be closely linked to age at diagnosis, being distinctly uncommon in boys younger than 10 years, and being present in up to 50% of older boys.
98,99 Alveolar histology is distinctly unusual in sarcomas of the genitourinary tract. Several recent analyses have raised the question of whether select patients with group I alveolar histology in this location may have a favorable outcome with more limited therapy.
100,101
Extremities
Sarcomas of the extremity are characterized by swelling in the affected body part (
Fig. 31.5). The male-to-female ratio is approximately 1:1. Pain, tenderness, and redness may occur. Between half and three-quarters of these tumors are alveolar.
102,103 Regional lymph node spread may be found in 20% to 40% of patients undergoing surgical exploration and is more likely if the primary tumor is an ARMS than an ERMS.
102,103,104,105 The tumors can be extensive because of their propensity to spread along fascial planes. The fact that injuries are frequent and expected on the extremities of school-aged children may lead to a delay in diagnosis; however, early dissemination to regional nodes and/or distant sites is often seen by the time the tumor is large enough to come to clinical attention.
Trunk
Truncal sarcomas are similar in evolution to those of the extremities, in that they exhibit all histologic types and have a tendency for local recurrence despite wide local excision and for distant spread. They are of relatively large diameter compared with tumors of the head and neck or of the bladder.
106 Contiguous involvement of the thoracolumbar spine may exist, depending on the location of the primary lesion, but regional lymph node spread is unusual.
Other Sites
Intrathoracic and Retroperitoneal-Pelvic Regions
Intrathoracic and retroperitoneal-pelvic tumors can become large before the diagnosis is made because they are deep within the body.
107 They are often incompletely accessible to the surgeon, because vital vessels are usually surrounded, and wide infiltration is the rule; however, more recent data fail to support the notion that differences in outcome for patients with thoracic tumors are accounted for by a higher proportion of patients with unresectable disease.
108 Patients with tumors in these locations have a higher-than-expected risk of local recurrence despite
combined-modality treatment. Aggressive attempts at initial or delayed surgical resection, combined with appropriate postoperative radiotherapy, may improve prognosis.
109
Perineal-Perianal Region
Lesions in the perineal-perianal region are unusual.
109 They can mimic abscesses or polyps. They tend to arise more commonly in adolescents and frequently present with unfavorable characteristics including alveolar histology (especially in adolescents), large tumor size, and regional nodal involvement.
110
Biliary Tract
Biliary tract tumors are substantially more rare than perineal-perianal tumors. They often produce obstructive jaundice, spread within the liver, and then spread to the retroperitoneum or lungs.
111Aggressive surgical resection appears to be less important to good outcome for tumors in this location.
112
Less Common Sites
Occasionally, the liver, brain, trachea, heart, breast, or ovary may harbor a primary sarcoma.
113,114,115,116,117 In some cases, no definite primary site can be determined.
118
STAGING
Assessing the extent of the tumor in every patient is critical because therapy and prognosis depend on the degree to which the mass has spread beyond the primary site. Patients with localized, surgically removable tumors have a better prognosis than do those whose disease has produced clinically detectable metastatic deposits. Two major staging systems are currently employed in combination: a surgicopathologic staging system known as Clinical Group (CG), developed by the Intergroup Rhabdomyosarcoma Study Group (IRSG) in 1972 (
Table 31.1), and the pretreatment, site-modified TNM staging system (Stage), developed by the IRSG in the late 1990s (
Table 31.2).
133 CG defines patients by the extent of their initial surgery, with subclassification of patients with microscopic residual disease (group II) with (II B, C) or without (II A) regional N1. The TNM system, which was retrospectively evaluated by numerous investigators and shown to be highly predictive of outcome, divides patients into favorable and unfavorable sites, and requires “upstaging” of patients with unfavorable-site tumors that are large (greater than 5 cm) and/or have clinical evidence of regional N1.
133,134,135,136 Favorable sites include the orbit and eyelid, and other non-PM head and neck structures, as well as nonbladder, nonprostate genitourinary locations (paratesticular, vulva-vagina-cervix-uterus). All other primary sites are considered unfavorable and include the extremities (including the buttocks and perineum), urinary bladder and prostate, cranial PM sites, and the trunk and retroperitoneum.
The likelihood of infiltration of regional lymph nodes or adjacent structures varies with the site of the primary tumor, ranging from as low as 5% for head and neck tumors to as high as 50% for extremity and paratesticular tumors in boys 10 years and older.
83,102 Imaging studies, particularly PET scan, and physical examination findings are usually adequate for establishing the presence of regional N1. If staging or local or systemic treatment options will be affected, consideration should be given to surgical removal of palpably or radiographically enlarged regional lymph nodes. Routine surgical sampling of radiographically “benign” regional nodes is unwarranted with two important exceptions: all
cases of extremity RMS should undergo aggressive sampling of regional nodal basins, and older boys (10 years of age and older) with paratesticular tumors should undergo ipsilateral retroperitoneal lymph node dissection (iRPLND). Radiation therapy (RT) is delivered to the region if tumor involvement of nodes is found on pathologic examination. Although there is no deleterious impact of N1 on prognosis for patients with ERMS, there is a dramatic adverse prognostic significance of regional NI for patients with ARMS with a magnitude of difference in 5-year failure-free survival (FFS) (43% vs. 73%) that is even greater than the difference in outcome based on a solitary site of metastatic disease (23%).
137Radiographic studies and bone marrow examination are used to define the anatomy of the primary tumor and to ascertain whether regional nodal and/or distant metastases are present; histologic verification of radiographic abnormalities is not required; however, where treatment issues will be determined by the presence or absence of regional or distant metastatic disease, surgical evaluation of an equivocal radiographic abnormality may be warranted. Histologic subtype does not affect either the Stage or the CG, although it may impact systemic and/or local treatment choices (e.g., the use of local irradiation in completely resected [group I], low stage [I and II] ARMS).
138 The classification system and assignment to treatment protocol can seem complicated (see
Table 31.3); a Web-based decision support tool can aid in the process of appropriate treatment assignment.
139