Rhabdomyosarcoma
 ANATOMY
ANATOMY
Rhabdomyosarcoma (RMS) is a highly malignant soft tissue sarcoma that arises from unsegmented, undifferentiated mesoderm or myotome-derived skeletal muscle. It may occur at any site in the body, but the most frequently involved sites are the orbit, 9%; head and neck (excluding parameningeal tumors), 7%; parameningeal, 25%; genitourinary 31%; extremity, 13%; trunk, 5%; retroperitoneum, 7%, and other sites 3%.1
Epidemiology and Risk Factors
RMS is the most common of the childhood soft tissue sarcomas, with an annual incidence of 4.4 per 1 million whites and 1.3 per 1 million blacks. The male-to-female ratio is ~1.5 to 1.0, and males may have slightly better overall survival.2
The great majority of patients are <10 years of age at the time of diagnosis, and approximately 5% are <1 year of age. There are two peak age frequencies, at ages 2 to 6 and in adolescence. Tumors in the younger age group are likely to be of embryonal histology (or one of its subtypes). About 25% of patients are ≥10 years at diagnosis and their tumors are more commonly of alveolar histology. Age has been identified as an independent predictor of prognosis, with children <1 year and >10 years having inferior survival.3 Adults with RMS have been reported to have poor outcomes, although there is evidence that when treated aggressively using pediatric-type protocols, the prognosis may be similar to that of younger patients.4,5
The cause of RMS is unknown; however, it is associated with several environmental exposures including paternal cigarette use, prenatal x-ray exposure, and maternal recreational drug use.2 RMS is also associated with disorders in development, including central nervous system, genitourinary, gastrointestinal, and cardiovascular anomalies, and with congenital disorders including congenital pulmonary cysts, Gorlin basal cell nevus syndrome, and neurofibromatosis. In addition, RMS is the most frequently occurring childhood cancer in families with Li-Fraumeni syndrome, and its incidence is increased in children with neurofibromatosis type 1, Beckwith-Wiedemann syndrome, and Costello syndrome.
Recent developments in cytogenetics and molecular genetics now provide a more comprehensive understanding of the origin and biologic behavior of RMS. These are discussed in more detail later.
 NATURAL HISTORY AND PATTERNS OF SPREAD
NATURAL HISTORY AND PATTERNS OF SPREAD
There are unexplained associations of site of primary tumor with age at diagnosis and tumor histology. For example, tumors arising in the urinary bladder and vagina occur primarily in infants and often are of the embryonal or botryoid histologic type. Tumors arising in the trunk and extremity occur in adolescents and are often alveolar or undifferentiated type. Tumors of the head and neck area occur throughout childhood and are commonly of the embryonal type.
RMS, a locally invasive tumor often with a pseudocapsule, has the potential for local spread along fascial or muscle planes, lymphatic extension, and hematogenous dissemination. The overall risk of regional lymphatic spread is approximately 15%, but varies with the site of the primary lesion. Lymph node metastases are rare in orbital tumors, but they occur in approximately 15% of tumors at other head and neck sites, most commonly the nasopharynx. Accounting for staging inaccuracies, regional lymph node extension occurs in approximately 25% of children with paratesticular, extremity, and truncal tumors.6 The risk for lymph node involvement also correlates with primary tumor invasiveness and large tumor size.
Hematogenous metastases are detected at the time of presentation in approximately 15% of patients, particularly those with truncal and extremity primary tumors. The most common sites of hematogenous dissemination are lungs, bone marrow, and bone. Malignant pleural and peritoneal effusions may also accompany tumors primary to the chest and abdomen or pelvis, respectively.7
 CLINICAL PRESENTATION
CLINICAL PRESENTATION
Because RMS occurs in multiple primary sites, there are many site-specific clinical signs and symptoms. It usually presents, however, as an asymptomatic mass. When symptoms are present, they relate to mass effect on associated organs and tissues. Tumors of the orbit may cause proptosis and ophthalmoplegia. Patients with parameningeal tumors often present with nasal, aural, or sinus obstruction, cranial nerve palsy, and headache. Genitourinary tumors may cause hematuria, urinary obstruction, or constipation.
 DIAGNOSTIC WORKUP
DIAGNOSTIC WORKUP
Determination of tumor extent is best done with a multidisciplinary approach by a radiation oncologist, pediatric oncologist, and appropriate subspecialty surgeon. An expeditious local and systemic workup is essential because these tumors have the potential to grow rapidly. The initial assessment by all members of the team permits accurate staging and the formation of a uniform treatment plan. Table 87.1 provides recommendation for diagnostic workup at various sites. Early experience with combined positron emission tomography (PET) and computed tomography (CT) scanning indicates this modality may be a valuable component of staging8 and may in fact provide more accurate staging than conventional imaging.9 PET-CT may be especially valuable in assessing response to therapy, as conventional imaging has not shown a good correlation of tumor response to long-term clinical outcome.10,11 Some children whose tumors exhibit characteristic fusion transcripts can be shown to have micrometastatic disease using reverse-transcriptase polymerase chain reaction techniques, even when there is no evidence of metastases from routine diagnostic procedures. The clinical significance of this is, however, unknown.12
 STAGING SYSTEMS
STAGING SYSTEMS
The clinical grouping classification used extensively by the Intergroup Rhabdomyosarcoma Study Group (now known as the Children’s Oncology Group Soft Tissue Sarcoma [COG STS] committee) investigators is somewhat of a misnomer because it actually requires surgical pathologic evaluation (Table 87.2). It is not a staging system and does not accurately reflect the biology of the disease, rather it reflects the surgical procedure selected for an individual patient. It is, however, useful for guiding decisions for radiotherapy, based on the amount of residual tumor after the initial surgical procedure. A more valid pretreatment staging system uses a TNM (tumor, node, metastasis) approach, which emphasizes characteristics of the primary tumor, size and invasiveness, nodal status, and systemic spread. Noninvasiveness, small size, and an absence of metastases have been shown to influence prognosis.13 The site of primary tumor also has a significant impact on survival.14,15 The Intergroup Rhabdomyosarcoma Study IV (IRS-IV) has prospectively demonstrated the validity of a staging system incorporating TNM classification along with primary tumor site (Table 87.3). Three-year failure-free survival was 86% for stage 1 tumors, 80% for stage 2, 68% for stage 3, and 25% for stage 4.1,7 Of note, the clinical grouping did not correlate as well with survival. In fact, failure-free survival for clinical group II patients in the IRS-IV study was superior to clinical group I patients (86% vs. 83%), probably reflecting the routine use of radiotherapy for group II patients.
TABLE 87.1 RECOMMENDED WORKUP FOR TUMORS AT VARIOUS SITES
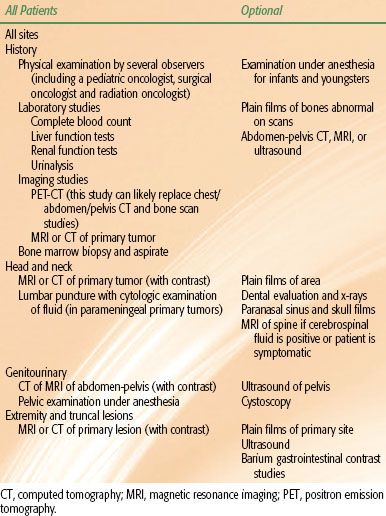
TABLE 87.2 INTERGROUP RHABDOMYOSARCOMA STUDY CLINICAL GROUPING CLASSIFICATION

TABLE 87.3 INTERGROUP RHABDOMYOSARCOMA STUDY PRETREATMENT STAGING SYSTEM

TABLE 87.4 INTERNATIONAL CLASSIFICATION OF RHABDOMYOSARCOMA

 PATHOLOGIC CLASSIFICATION
PATHOLOGIC CLASSIFICATION
The histogenesis of RMS can be traced from mesoderm to mesenchyme and ultimately to striated muscle tissue. The classification of RMS initially used by the IRS investigators consisted of four histologic subtypes: embryonal, botryoid subtype of embryonal, alveolar, and pleomorphic. Embryonal histologies comprise approximately two-thirds of all cases, with most of the rest having alveolar histology.16 Other variants including a “solid” alveolar pattern, considered a subtype of alveolar RMS, a spindle cell subtype of embryonal RMS, and a diffuse anaplastic variant have also been described.17
To improve reproducibility of pathologic subtyping and prognostic utility, pediatric pathologists developed an updated classification system: the International Classification of Rhabdomyosarcoma. This system is based on a review of IRS-II data and it groups pathologic subtypes into distinct prognostic groups (Table 87.4). The International Classification of Rhabdomyosarcoma system appears to be predictive of outcome and has been reproduced by several reference pediatric pathologists.17,18 The superior prognosis group, comprising of two subsets (botryoid and spindle cell), carries a projected 5-year survival rate of 88% to 95%.18 The botryoid subtype, a polypoid variant of embryonal RMS, has a grapelike appearance. The stroma consists of loose cellular tissue with a myxoid appearance. Under the superficial stroma is a hypercellular zone of tumor cells called the cambium layer of Nicholson. Botryoid tumors are usually noninvasive and localized and occur in mucosal-lined organs such as the vagina, urinary bladder, middle ear, biliary tree, and nasopharynx. The spindle cell subtype of embryonal RMS has a spindled appearance, often with a storiform pattern. It is frequently found in paratesticular sites.
Patients with embryonal RMS have an intermediate prognosis, with an 83% failure-free survival at 3 years.14 The embryonal type consists of blastemal mesenchymal cells that tend to differentiate into cross-striated muscle cells. There is often a considerable variation in degree of cytoplasmic development, ranging from primitive mesenchymal to highly differentiated muscle tumor cells. Most of the tumor cells have eosinophilic cytoplasm, which is positive by periodic acid Schiff staining. Immunohistochemistry may demonstrate actin- or desmin-positive reactions. Ultrastructural studies exhibit evidence of myogenesis with the presence of thick and thin cytoplasmic intermediate filaments or Z-band material. Ribbon or strap-shaped cells and tadpole cells are characteristic. The presence of cross-striations confirms the diagnosis. The embryonal form may be distinguished from the other subtypes by specific structural abnormalities. A consistent loss of heterozygosity at the chromosome 11p15.5 locus suggests that this site is specific for the embryonal subtype,19 although, unlike alveolar histology, no characteristic translocation has been identified. Immunohistochemical presence of epidermal growth factor receptor and fibrillin-2 appears to be highly specific for embryonal histology and is predictive of a favorable outcome.20,21 Dysregulation of the RAS pathway has also been found in some patients and may be involved in the pathogenesis of this subtype.22 The embryonal histology is found most commonly in the orbit, head and neck, and genitourinary sites.
The group with poor prognosis includes alveolar, diffuse anaplastic, and undifferentiated sarcomas. With routine use of immunohistochemistry and molecular genetic analysis, about one-third of RMSs are now classified as alveolar; it is most commonly found in adolescents with truncal, retroperitoneal, and extremity tumors. The alveolar subtype is characterized by a pseudoalveolar pattern of connective tissue trabeculae, lined by large rhabdomyoblasts and multinucleated giant cells. The “solid” variant of alveolar RMS grows as solid nests of closely aggregated tumor cells with less alveolar pattern. Alveolar histology is strongly associated with hyperdiploid content.23 Approximately 75% of children with alveolar RMS exhibit a characteristic translocation involving chromosomes 2 and 13, t(2;13)(q35;q14) and occasionally a 1;13 translocation.19,24 These translocations correspond to abnormal fusion genes involving PAX3-FKHR and PAX7-FKHR, respectively, and are probably the initial oncogenic events in these tumors.25,26 Immunohistochemical presence of AP2-β and P-cadherin is also highly specific for alveolar histology.20,21 The projected 3-year failure-free survival for children with the alveolar subtype is 66%.1 Recent data suggest that fusion-negative alveolar RMS is more similar to embryonal histology, both clinically and molecularly, and may be more appropriately managed with those treatment algorithms.27
Undifferentiated sarcoma is largely a diagnosis of exclusion; it consists of a diffuse cell population of primitive, noncommitted mesenchymal cells. The 3-year failure-free survival rate of patients with undifferentiated sarcoma is 55%.1 Today, sarcomas that lack characteristics of differentiation are most appropriately managed as non-RMS soft tissue sarcomas.
The pleomorphic type is extremely rare; many cases formerly classified as pleomorphic RMS are currently considered to be malignant fibrous histiocytoma. Previously, tumors classified as extraosseous Ewing’s sarcoma were treated using guidelines for RMS. These are more appropriately considered in the Ewing family of tumors and are managed as such.
 PROGNOSTIC FACTORS AND THERAPEUTIC CONSIDERATIONS
PROGNOSTIC FACTORS AND THERAPEUTIC CONSIDERATIONS
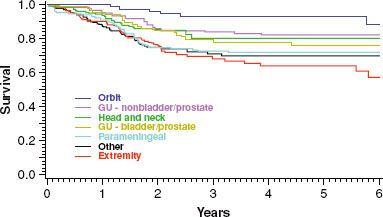
Because RMS is protean in presentation, factors such as age, site, stage, extent of disease, and pathologic characteristics of the tumor influence therapeutic decisions. These prognostic factors are interrelated and are best discussed as a function of the specific site (Fig. 87.1). Although most treatment failures occur within 3 years of diagnosis, about 10% of children who are free of disease at 5 years will subsequently experience disease recurrence.28
FIGURE 87.1. Survival curves for 883 children with nonmetastatic disease entered onto the fourth Intergroup Rhabdomyosarcoma Study are shown by anatomic site of the primary tumor. GU, genitourinary; B/P, bladder-prostate. (Adapted from Crist WM, Anderson JR, Meza JL, et al. The Intergroup Rhabdomyosarcoma Study-IV: results for patients with non-metastatic disease. J Clin Oncol 2001;19:3091–3102.)
Orbit
The orbit has long been recognized as a favorable prognostic site. In addition to prompt recognition of the tumor, the paucity of lymphatics in this area means that lymphatic extension is rare. Most tumors in this site have embryonal histology, and hematogenous metastasis at the time of diagnosis is uncommon. Approximately 10% are alveolar histology, however, and the prognosis for these children is more guarded.29
When treatment for orbital tumors is individualized, it is generally agreed that no surgical procedure should be used that may compromise vision. In most patients, this means that biopsy only should be performed to provide the diagnosis. Primary treatment typically consists of vincristine, actinomycin-D, and cyclophosphamide (VAC) or vincristine and actinomycin-D (VA) chemotherapy with local radiotherapy beginning between the 3rd and 12th week of treatment. Radiation doses of approximately 50 Gy are often used, although results from the IRS-V study suggest that 45 Gy may be sufficient when given with a cyclophosphamide-containing chemotherapy combination.30,31 Using this approach, cure rates of >90% can be achieved.14 Chemotherapy without irradiation has resulted in local relapse and inferior event-free survival.32 Although salvage radiotherapy for these patients can still be curative, functional vision in this setting is often poor.33 Orbital exenteration should be reserved for salvage treatment and enucleation for management of posttreatment ocular complications.
With a combined-modality approach, radiotherapy can be directed to the tumor plus a margin without necessarily irradiating the entire orbit. Technique is very important for minimizing corneal and lacrimal gland dose and for preserving useful vision, ocular function, and appearance. Photon irradiation with the eyelid open can minimize the corneal dose when an anterior field is used and may be associated with improved long-term functional outcome.34 Three-dimensional conformal or intensity-modulated radiotherapy technique is optimal for treating the target volume and sparing normal structures, and proton radiation has also been used successfully.35
FIGURE 87.2. This 12-year-old girl was treated at age 3 years for an embryonal rhabdomyosarcoma of the nasal cavity. Her treatment consisted of excisional biopsy, followed by VAC (vincristine, actinomycin-D, and cyclophosphamide) chemotherapy and 41.4 Gy radiotherapy. Slight hypoplasia of the midface is evident, but overall cosmesis is excellent.
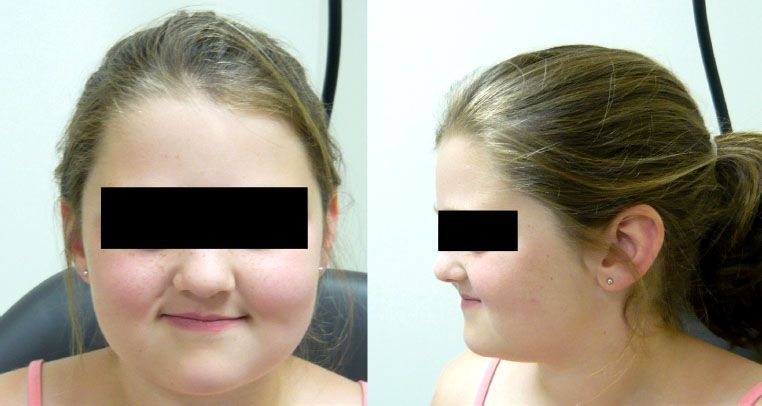
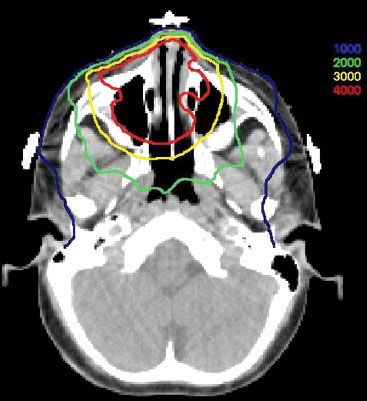
FIGURE 87.3. Isodose distribution from the treatment of the child in Figure 87.2 (doses in cGy).
Head and Neck: Parameningeal Sites
Nonorbital RMS of the head and neck is grouped into parameningeal sites (nasopharynx, nasal cavity, paranasal sinuses, middle ear, pterygopalatine fossa, and infratemporal fossa) and nonparameningeal sites based on differences in natural history, treatment, and prognosis. Parameningeal RMS represent the majority of nonorbital head and neck RMS, and radiotherapy is essential for maximizing the chance of cure.14,36 These tumors have a propensity for invading the base of the skull, creating a potential for cranial nerve palsy and direct extension into the central nervous system, a pattern of spread that is seen in as many as 41% of these patients.37 Historically, as many as 35% of children with tumor arising in a parameningeal site would later have meningeal extension, and previous irradiation regimens called for whole-brain irradiation as part of central nervous system prophylaxis.38 However, the prognosis of these patients is markedly improved with appropriate imaging, multiagent chemotherapy, and adequate irradiation of the primary tumor and adjacent meninges, and studies demonstrate that whole-brain irradiation is not necessary, even in the presence of direct intracranial tumor extension.39 Patients with known meningeal dissemination should receive craniospinal irradiation. A radiation dose of 50.4 Gy in 28 fractions to the primary site is commonly used. Data from the IRS studies show improved local control in patients with intracranial tumor extension when radiotherapy is started within 2 weeks of diagnosis,37 although other reports show no disadvantage with delayed radiotherapy.40
Aggressive surgery is rarely indicated because complete resection usually is not possible, does not obviate the need for high-dose radiation therapy, and often results in a delay of systemic chemotherapy because of postoperative complications. Surgical approaches to these tumors have been described by proponents of multispecialty skull-base surgery, but the efficacy of these approaches has not been firmly established.41 Delayed surgical resection has been proposed as beneficial for children with residual tumor after completing chemotherapy and radiotherapy but is not considered standard of care.42,43 The role of postradiation surgical resection is being investigated in select intermediate-risk patients enrolled onto IRS-V.
Five-year survival for patients with parameningeal RMS is approximately 75% with adequate radiotherapy.44 For the subset of parameningeal tumors arising in the nasopharynx or nasal cavity, middle ear, and parapharyngeal locations, survival may be even higher,39 and functional and cosmetic outcome can by good in spite of an aggressive treatment regimen (Figs. 87.2 and 87.3).
Head and Neck: Nonparameningeal Sites
Children with tumors in nonparameningeal head and neck sites tend to have a better outcome than their parameningeal counterparts (80% 5-year failure-free survival in IRS-IV) and require less-intensive chemotherapy.1,14 These sites include the scalp, parotid, oral cavity, larynx, oropharynx, and cheek. These tumors may be more amenable to complete gross surgical excision compared with their parameningeal counterparts. Approximately 15% of these patients present with regional lymph node metastases. Radiotherapeutic management is based on the amount of residual tumor after surgery. Draining regional lymph nodes are not routinely irradiated unless they are considered to be involved with tumor by clinical or pathologic assessment.
Pelvis
Pelvic tumors usually are divided into anatomic subgroups because the natural history, treatment, and prognosis are different for each site. Some children present with locally advanced pelvic tumors for which an exact site of origin cannot be determined. These large tumors are associated with an unfavorable prognosis.
Bladder and Prostate Tumors
Bladder and prostate primary tumors account for about half of all pelvic RMS36; 75% of patients are age <5 years at presentation, and more than 90% of these tumors are of the embryonal histologic subtype, with approximately one-third having a botryoid morphology. In boys, it is often difficult to differentiate a tumor of prostatic origin from one of bladder origin because disease usually involves both structures. However, patients with tumors arising in the prostate have significantly inferior survival compared with those with tumor confined to the bladder.45
Historically, anterior pelvic exenteration (or partial cystectomy for small tumors arising from the dome of the bladder) combined with chemotherapy and irradiation for microscopic or gross residual disease has been associated with a survival rate of approximately 70%.15 More recently, emphasis has been on limited surgery to preserve bladder function. The IRS-II study treated these patients primarily with chemotherapy followed by delayed surgery or radiation therapy when there was residual or recurrent disease. This approach resulted in an inferior 3-year disease-free survival rate of 52%, compared with 80% for a primary radical surgical approach and only 22% of patients survived with intact bladders.46 Subsequently, IRS-III intensified the therapy with systematic use of planned irradiation 6 weeks after the start of treatment and added cisplatin and doxorubicin (Adriamycin) chemotherapy.47 The 5-year survival rate for patients with locoregional disease was 82%, with 64% of surviving patients retaining functional bladders, demonstrating the necessity of routine radiation therapy early in treatment.48 Survival of patients with locoregional bladder or prostate tumors treated on IRS-IV was similar, with 40% of all enrolled patients reporting normal bladder function at follow-up.49 A more recent analysis showed maintenance of continence in 69% treated with conservative surgery in a multimodality treatment program.50
The German Cooperative Weichteilsarkom Studiengruppe CWS-96 protocol treated children with bladder or prostate RMS using multiagent chemotherapy followed by response-adapted radiotherapy and surgery. Children with complete resection did not have radiotherapy, while others received 32 to 44.8 Gy using 1.6 Gy twice a day, depending on the tumor’s IRS group and response to chemotherapy. Five-year event-free survival was 70%.51
A pooled analysis of patients treated prospectively on several cooperative group trials from North America and Europe showed improved local control for those who received radiotherapy as part of their initial treatment, but this did not translate into improvement in overall survival.52
Paratesticular Tumors
Paratesticular tumors represent approximately 7% of all RMS and may arise anywhere along the spermatic cord, from the intrascrotal area through the inguinal canal.53 At presentation, the tumor usually is a painless scrotal or inguinal mass that does not transilluminate. Most boys with paratesticular RMS present with early-stage disease that is amenable to complete resection and is associated with cure rates approaching 90%. As with prostatic primary tumors, the lymphatic network is rich, draining directly to the retroperitoneal nodes along the external iliac and spermatic vessels, the aorta, and the vena cava. The incidence of retroperitoneal lymph node involvement varies with the age of the patient and method of staging (surgical vs. nonsurgical). In the IRS-III study, retroperitoneal lymph node sampling was done in most patients, showing a 14% incidence of node involvement for children <10 years of age and a 47% incidence for those ≥10 years. In IRS-IV, thin-cut CT without surgical sampling was used for staging, and the incidence of detected nodes dropped to 4% and 13% for the two age groups, respectively.53
The recommended surgical procedure for the primary tumor is inguinal orchiectomy. If there is no evidence of invasion into the scrotum and the proximal spermatic cord is free of tumor, this procedure is considered equivalent to an amputation, and no further local therapy is necessary. Surgical staging of retroperitoneal lymph nodes is controversial. European investigators do not recommend retroperitoneal lymph node sampling for these patients, preferring to treat with intensified chemotherapy in high-risk patients and salvage radiotherapy, if necessary.54,55 In the IRS, the high risk of nodal involvement in certain subsets of these patients has led to the recommendation for ipsilateral retroperitoneal nerve-sparing node dissection for staging of all children ≥10 years of age.53 In the absence of histologic documentation, enlargement of retroperitoneal lymph nodes on thin-section CT imaging is considered evidence of tumor involvement.
Patients with nodal disease have a 5-year survival rate of 69%, compared with 96% for those without regional nodal disease (P <.001). Regional lymph node irradiation to the periaortic and ipsilateral iliac nodes is recommended when there is nodal involvement.56 Surgical violation of the scrotum or tumor extension to the structure is an indication for hemiscrotectomy or, less commonly, scrotal irradiation. If scrotal irradiation is used, orchiopexy should be considered prior to treatment to protect the remaining testes. Treatment programs must be planned to reduce morbidity, particularly among this group with high likelihood of cure.
Gynecologic Tumors
Tumors arising in the vulva, vagina, cervix, and uterus are about one-third as common as bladder and prostate primary tumors and account for 4% of all RMS.57 Within this group, the vagina is the most common site of origin.
Patients with vaginal tumors are often much younger than those with other pelvic RMS, with most girls diagnosed before the age of 3 years. Most present with a vaginal mass or discharge; botryoid morphology is common. Initial surgery is used primarily for diagnosis, although gross tumor resection is occasionally possible without cosmetic or functional deformity. These tumors are often quite sensitive to chemotherapy, and treatment regimens have previously used a strategy of chemotherapy only, reserving surgery and radiotherapy for persistent or recurrent tumor.57–59 However, analysis of data from the IRS-IV and IRS-V studies have revealed high rates of local failure for vaginal tumors when local control with surgery or radiotherapy is omitted.31,60 Current guidelines from the COG STS committee call for radiotherapy in all patients with postsurgical microscopic or macroscopic tumor.
Uterine, cervical, and vulvar tumors receive chemotherapy and radiotherapy based on the amount of tumor present after initial surgery. Intracavitary and interstitial brachytherapy are useful irradiation techniques in some of these patients.61 Data are limited, but with proper patient selection, disease control is excellent and late normal tissue effects may be significantly less with brachytherapy than is seen with external-beam techniques.62 Permanent implants with iodine-125, temporary low–dose-rate, and high–dose-rate brachytherapy have all been used, and there are no clear differences among these techniques in terms of disease control or late effects. When temporary implants are used, high–dose-rate brachytherapy has the practical advantage of minimizing radiation exposure to the family and medical personnel caring for the child.
Survival after treatment is excellent. Children ages 1 to 9 years have a 98% 5-year survival. Survival for infants and adolescents approaches 90%, although these patients may require more intensive systemic therapy for cure.57
Other Pelvic Sites
These tumors include perianal, perirectal, and perineal primary sites. Regional lymph node involvement is relatively common. The location of these primary tumors creates surgical and irradiation challenges. Combined chemotherapy and radiation therapy programs are favored over primary surgical procedures if excision requires exenteration with urinary and fecal diversion procedures or is associated with organ or sphincter dysfunction.
Extremity
Tumors arising in the extremity are often of the alveolar or undifferentiated subtypes, large, deeply invasive, and associated with a high probability of lymphatic and hematogenous metastasis.63 Complete surgical resection is difficult to achieve, usually requires extensive dissection, and is associated with a high risk of residual disease. Because radiation therapy and multiagent chemotherapy have been shown to provide excellent local control, it is advisable to avoid disfiguring and mutilating surgical procedures, with their attendant functional disabilities, and to recommend limb-salvage procedures including irradiation and chemotherapy.
Regional lymph node involvement is present in approximately 24% of patients and confers a more guarded prognosis.63 Aggressive surgical staging of draining lymph node basins is important to define extent of involvement, although if lymph node dissection is performed, it is for the purpose of staging, not for treatment. Sentinel lymph node mapping and biopsy are being investigated for their diagnostic and prognostic value.64,65 Radiotherapy of involved regional lymphatics is mandatory, and aggressive treatment of in-transit nodal sites may improve overall local control.66
Radiation therapy for extremity primary tumors requires careful immobilization techniques, sparing of nonirradiated skin for lymphatic drainage, and use of shrinking fields. Routine physical therapy during and after radiation therapy is important for obtaining an optimal functional result. Overall survival at 3 years for these patients is about 70%, although failure-free survival is only 55%.67
Other Sites
Patients with tumors arising in sites such as a paraspinal, retroperitoneal, or intrathoracic location have a poor outcome compared with other patients.14 Most patients with tumors in these locations are unable to undergo complete resection of the tumor. Both local and distant relapse are common. These patients should be treated aggressively with high-dose radiation therapy and multiagent chemotherapy.
Metastatic Disease
Hematogenous or distant lymph node metastasis at the time of diagnosis is an ominous finding, although not all these children do poorly. The subset of patients who are <10 years of age and have only one or two sites of metastatic disease may have long-term survival chances of >50%.7 Intensive multiagent chemotherapy plays a major role in the treatment of these patients, although marrow ablative techniques have not improved efficacy compared with conventional chemotherapy approaches.68,69 Local control of the primary tumor is site specific, as previously described. Metastatic sites should be treated with radiotherapy when feasible.
 GENERAL MANAGEMENT
GENERAL MANAGEMENT
A multidisciplinary approach using surgery, irradiation, and chemotherapy is important in the management of RMS; however, the optimal sequence and specific application of each modality continue to be investigated. Although the primary goal remains long-term cure, improvement in therapeutic results necessitates considerations of quality of life, with particular attention to maximization of functional and cosmetic results.
Surgery
Before the era of multidisciplinary therapy, surgical ablation resulted in a long-term survival rate of approximately 20% of those patients able to undergo resection.70 Certain primary sites represented exceptions to these data; for example, approximately 50% of those with localized disease of the orbit survived after orbital exenteration.71 However, with the introduction of effective adjunctive treatment, preservation of function and appearance became major goals. The concept of reasonable surgery has evolved. It involves removal of the bulk of tumor with maximal conservation of anatomic structures, including preservation of bladder, bowel, and sexual function in patients with tumors of genitourinary origin; limb function in patients with extremity tumors; and vision, voice, deglutition, and appearance in patients with head and neck tumors.
Resection of RMS from normal surrounding tissues is often technically challenging. Only 20% of tumors are located in sites where complete excision can readily be accomplished without an undesirable loss of function or cosmesis.14 An additional 20% of patients have compromised surgical procedures, leaving microscopic residual disease. Sixty percent of patients have tumors amenable to biopsy only or present with metastatic disease and are not candidates for primary resection. When the IRS surgical grouping system is used, patients with tumor amenable to complete excision fare better than those who have subtotal resection or biopsy alone. However, the tumors that are most accessible to surgical excision are small and noninvasive and are confined to the organ or structure of origin. Assessment using a TNM system demonstrates that prognosis is dictated by tumor size and invasiveness rather than by the initial surgical approach.72 Furthermore, combined-modality therapy provides good local control of the primary tumor, even after subtotal excision.73 For these reasons, the trend has been toward less-aggressive surgical resection, with more reliance on radiation therapy and chemotherapy to provide local control.14
In cases of suspected RMS, the initial surgical procedure should be an incisional biopsy. Surgical excision is indicated if it can be done without compromise of function or cosmesis. Normal tissue margins of at least 5 mm around the tumor are usually required to consider the resection complete (IRS group I), although this is sometimes not feasible in some anatomic sites and smaller margins may suffice. If microscopic disease remains after initial resection, a primary re-excision can be considered prior to beginning chemotherapy. Those children who can be rendered microscopically free of disease by this procedure have an improved outcome, compared with children who remain in clinical group II following initial surgery.74,75 Amputation of an extremity, orbital exenteration, mutilating surgery of the head and neck area, therapeutic lymphadenectomy, and radical neck dissection are procedures reserved in case initial therapy fails.
Second-look operations (also termed delayed primary excisions) may be useful for converting partial responses after chemotherapy into complete responses, and there is evidence that these procedures may improve survival.43 However, data regarding efficacy of second-look operations are mixed, with some finding no benefit with this procedure.10 To investigate if second-look surgery might allow a reduction in the amount of radiotherapy that is necessary to provide local tumor control, the IRS-V study evaluated this approach; preliminary results suggest that only select primary sites are appropriate for this approach, and final results are currently pending. Some investigators have used second-look operations in an attempt to eliminate radiotherapy, although this approach has resulted in inferior local control and survival.76 Second-look operations may be used to evaluate therapeutic response after chemotherapy or radiation therapy. In the IRS-III study, 28% of patients categorized as having clinical partial response and 43% of those scored as having no response to induction chemotherapy were reclassified as having pathologic complete response after second-look operation.77 These children enjoyed a survival rate similar to that of children who were able to undergo complete surgical excision at the time of initial diagnosis. Therefore, a clinical or radiographic evaluation indicating residual tumor after initial therapy may be misleading.
Chemotherapy
Chemotherapy is necessary in all cases. Several drugs have demonstrated single-agent activity measured as a percentage response rate, including vincristine (59%), dactinomycin (24%), cyclophosphamide (54%), cisplatin (15% to 21%), dacarbazine (11%), mitomycin-C (36%), etoposide (15% to 21%), ifosfamide (86%), irinotecan (23%), and topotecan (46%).78–80 Agents with known activity against central nervous system tumors, such as the nitrosoureas and methotrexate, have not shown activity against RMS.
The most extensive experience in combination chemotherapy is with VAC or VAC plus doxorubicin (VACA). Some studies have suggested that patients with embryonal histology tumors in favorable sites who have no gross residual disease or lymph node involvement after the initial surgical resection may be adequately treated with VA for 1 year, provided that irradiation is given for microscopic residual disease.14 However, data from the recently completed IRS-V study suggest that local control may be compromised when alkylating agents are omitted.30,31 Patients with unresectable pelvic tumors may benefit from the addition of doxorubicin and cisplatin to VAC, but tumors in other sites do not seem to benefit from the addition of these drugs compared with an intensive regimen of VAC alone.
Some subsets of patients, such as those with tumors of unfavorable histology or unfavorable site and those with extensive tumor burden, continue to fare poorly. Some of these patients with embryonal histology may benefit from intensification of the cyclophosphamide or ifosfamide component of their chemotherapy, although there are conflicting data regarding this.81,82 Patients with metastatic RMS benefit from the addition of ifosfamide and etoposide to the standard VAC regimen.83 High-dose chemotherapy with total-body irradiation and autologous bone marrow transplantation has not improved the outcome in these high-risk patients.68,69
Initial intensive chemotherapy has been used as a means of pharmacologic debulking, potentially allowing for a more conservative surgical approach or less-aggressive radiation therapy.46,84,85 However, response to induction chemotherapy—whether complete, partial, or no response—does not predict ultimate outcome.86 When chemotherapy alone is used for tumors in sites such as the head and neck or pelvis, most children require radiation therapy with or without a follow-up surgical procedure because of incomplete response or local recurrence.15,32,87,88 Omission of radiotherapy in these patients may result in inferior survival.76,89 Even patients with only microscopic disease after initial resection (group II) require radiotherapy to achieve optimal local control.90 In patients with group II disease who routinely receive radiotherapy, local control is 92%.91 The approach of initial chemotherapy followed by limited irradiation or less radical surgery may be appropriate in the management of infants and very young children, in whom late effects of aggressive surgery or high-dose, large-volume irradiation are particularly severe, although this may lead to inferior local control and survival.
Radiation Therapy
Adequate irradiation implies careful attention to volume and dose. It is essential to evaluate the soft tissue extent of the primary lesion by CT scan or magnetic resonance imaging. Because RMS tends to infiltrate tissue planes widely, tumors often extend beyond a fascial compartment and beyond the obvious visible margins. Careful examination by a radiation oncologist at the time of initial diagnosis, even if the treatment plan calls for neoadjuvant chemotherapy, is essential to establish the appropriate tumor volume.
Treatment portals are designed to encompass the involved region at the time of presentation (before chemotherapy) with margins that encompass surgical sites and biopsy tracts. A biopsy should be performed of clinically suspicious lymph nodes, or they should be included in the radiation therapy portal. Prophylactic lymph node irradiation is not necessary in children with clinically negative findings who will be receiving combination chemotherapy. The gross tumor volume is defined as the tumor as seen at the time of initial diagnosis. A clinical target volume of 1 cm is added and can be modified to account for anatomic barriers to tumor spread (such as the bony orbit in primary orbital tumors) or to account for regression of “pushing” the tumor border after chemotherapy, such as may occur in large pelvic tumors that initially displace contents of the peritoneal cavity. The planning target volume adds a patient-specific margin, which is typically about 5 mm. Many current treatment protocols use a cone-down boost or simultaneous integrated boost to any gross posttreatment tumor volume after a microscopic tumor dose has been delivered. Patients with tumors at parameningeal sites (middle ear, paranasal sinuses, nasopharynx, nasal cavity, infratemporal fossa, and parapharyngeal area) have developed meningeal extension of tumor when inadequate irradiation portals were used.39 Radiation therapy portals that cover the adjacent meninges in these patients can prevent meningeal relapse.14,37
Three-dimensional conformal and intensity-modulated radiotherapy treatment planning techniques are valuable for ensuring adequate treatment of the tumor volume and minimizing acute and chronic toxicity from the irradiation of uninvolved, adjacent structures.37,92–94 Proton-beam radiotherapy is being increasingly utilized as access to this form of therapy increases,35 and dosimetric studies consistently demonstrate decreases of integral dose to normal structures compared to photon-based techniques for many children with RMS (Fig. 87.4).95 The long-term clinical significance of these differences in integral dose has not been studied, but as the number of proton centers increases, many clinicians are recommending proton radiotherapy for most affected children. Others, however, express concern regarding the increased neutron dose associated with many current proton delivery technologies and urge caution before adopting protons as standard of care for RMS.96 Immobilization techniques that ensure reproducible portals are essential. Sedation or anesthesia may be necessary to ensure adequate implementation of the treatment plan. These complex programs are best conducted in regional centers by an experienced team of physicians, including a pediatric surgeon, pediatric anesthesiologist, pediatric oncologist, and radiation oncologist.
Radiation is necessary to ensure local tumor control in patients who are unable to undergo complete surgical resection. Local control of gross disease in most anatomic sites requires doses of 50 to 55 Gy. Data from the IRS-V D9602 study support a somewhat lesser dose of 45 Gy for gross tumor at orbital sites, especially if cyclophosphamide is included in the systemic therapy regimen.30,31 In the IRS-IV study, investigators studied the efficacy of a higher radiation dose, 59.4 Gy, given in 1.1-Gy fractions twice daily at 6-hour intervals for children with gross residual disease.97 This hyperfractionated regimen was compared in a prospective, randomized fashion to a standard radiotherapy regimen consisting of 50.4 Gy given as 1.8 Gy once daily. There was no difference in locoregional disease control, failure-free survival, or overall survival between the two groups. Therefore, the standard of care for group III RMS continues to be conventionally fractionated radiation with chemotherapy.
Ninety percent of patients with microscopic residual tumor achieve local control with 41.4 Gy, and the D9602 study results suggest that 36 Gy may be adequate for microscopically positive margins that are not associated with lymph node involvement. Investigators have been unable to generate a strict dose–response curve but have observed an association with age that suggests that lower doses, often given to infants and youngsters, are associated with higher relapse rates.98 They also suggest that local tumor control is greater for tumors <5 cm in diameter than for larger lesions, supporting the adult experience with soft tissue sarcomas.99,100
Results of the IRS-I study had indicated that radiotherapy was not needed for patients whose tumors were completely resected at diagnosis (group I). Subsequently, a reanalysis of data from the IRS-I to IRS-III studies suggested that the subset of group I patients with alveolar or undifferentiated histology had improved overall and failure-free survival when radiotherapy was given to the primary tumor site.101 A more recent analysis using data from IRS-IV did not, however, show a significant advantage for radiotherapy in group I alveolar tumors, and this practice is currently under review for the next generation of RMS protocols.102
Interstitial radiation therapy may play a role as primary treatment or as a boost after external-beam therapy for selected sites.41,62 The advantages of precise shaping of the dose distribution, sharp falloff of radiation dose, and shortening of overall treatment time are especially attractive in dealing with infants and young children. Some investigators report a decrease in late normal tissue effects when compared with external-beam techniques.103 There are no data regarding comparative efficacy or toxicity between high–dose-rate and low–dose-rate techniques. However, high–dose-rate remote brachytherapy may be particularly attractive in this patient population for logistical reasons. These children often require extensive care from family members and medical personnel during their treatment, and high–dose-rate techniques can eliminate radiation exposure to these caregivers.
The timing of radiation therapy must be carefully coordinated with planned surgical intervention and combination chemotherapy scheduling to optimize local control and ensure optimization of drug doses and unimpaired postoperative healing. Although radiation therapy is often delayed for several weeks to allow administration of neoadjuvant chemotherapy, some data suggest that earlier irradiation, particularly in high-risk patients, may provide better local tumor control and survival.37,104,105 Interaction between radiation and some of the commonly used chemotherapeutic drugs can produce undesirable early and late effects. This is particularly true of dactinomycin and doxorubicin. Radiation therapy given concurrently with these agents is usually avoided. In contrast, systemic treatment with drugs such as vincristine and cyclophosphamide can usually be continued concurrently with the administration of irradiation.
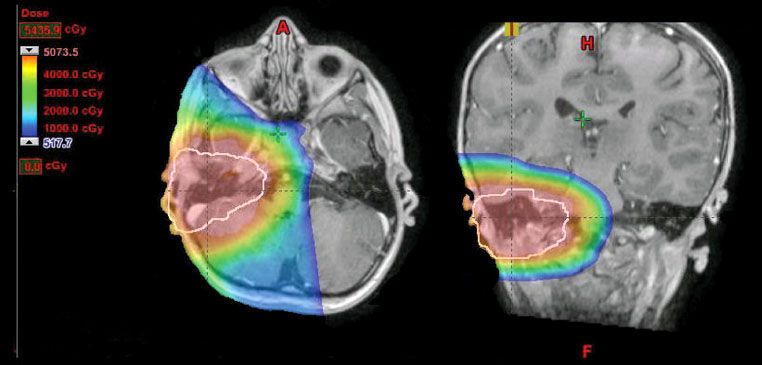
FIGURE 87.4. Isodose distribution in the axial and coronal planes for treatment of a middle ear RMS using protons. The physical properties of protons permit complete sparing of the contralateral structures and ipsilateral eye. (Courtesy of Danny Indelicato, MD.)