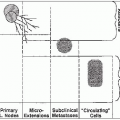
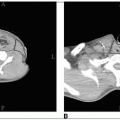
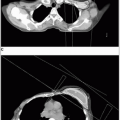
Rhabdomyosarcoma is a highly malignant soft tissue sarcoma that arises from unsegmented, undifferentiated mesoderm, or myotome-derived skeletal muscle.
Rhabdomyosarcoma is heterogeneous and arises in multiple sites (3).
Tumors of the head and neck area occur throughout childhood and are commonly embryonal.
Tumors arising in the trunk and extremity occur in adolescents and are usually alveolar or undifferentiated.
Tumors arising in the urinary bladder and vagina occur primarily in infants and often are embryonal or botryoid. This locally invasive tumor spreads along fascial or muscle planes and by lymphatic extension, and may have hematogenous dissemination.
Lymph node metastases are rare in orbital tumors but occur in approximately 15% of tumors at other head and neck sites (most commonly the nasopharynx), 25% of paratesticular tumors, and 20% of extremity and truncal tumors (2).
Hematogenous metastases are detected at the time of presentation in approximately 15% of patients. The most common sites of hematogenous dissemination are lungs, bone marrow, and bone (2).
Rhabdomyosarcoma usually presents as an asymptomatic mass. When symptoms are present, they relate to mass effect on associated organs.
Tumors of the orbit may cause proptosis and ophthalmoplegia.
Parameningeal tumors often present with cranial nerve palsy; headache; and nasal, aural, or sinus obstruction.
Genitourinary tumors may cause hematuria, urinary obstruction, or constipation (7).
The extent of the primary tumor is best determined with a multidisciplinary, expeditious workup by a radiation oncologist, pediatric oncologist, and appropriate subspecialty surgeon.
Recommended baseline evaluations are shown in Table 48-1 (7).
TABLE 48-1 Recommended Workup for Tumors at Various Sites | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
TABLE 48-2 IRS Clinical Grouping Classification | ||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||
Pretreatment staging uses a tumor-node-metastasis system, which emphasizes characteristics of the primary tumor, size and invasiveness, nodal status, and systemic spread.
In the IRS-IV study, disease is designated as stage I if the tumor is without hematogenous metastases and is in a favorable site, such as orbit, genitourinary non-bladder-prostate (paratesticular, vagina, vulva, uterus), or head and neck nonparameningeal.
Stage II tumors are in an unfavorable primary site such as bladder-prostate, extremity, parameningeal, and other sites and are smaller than 5 cm with negative regional lymph nodes.
Stage III tumors are in an unfavorable primary site, are larger than 5 cm and node negative, or are in any unfavorable site and node positive.
The classic classification of rhabdomyosarcoma, used by the IRS investigators, consists of four histologic subtypes: embryonal, botryoid subtype of embryonal, alveolar, and pleomorphic.
More recently, investigators have observed a subset of patients with a “solid” alveolar pattern, considered a subtype of alveolar rhabdomyosarcoma (31).
A lack of agreement among pediatric pathologists has led to the new International Classification of Rhabdomyosarcoma, based on a review of IRS-II data, which divides subgroups into distinct prognostic groups (Table 48-3).
The botryoid subtype, a polypoid variant of embryonal rhabdomyosarcoma, has a grapelike appearance; it is usually noninvasive and localized, and presents in mucosal-lined organs such as the vagina, urinary bladder, middle ear, biliary tree, and nasopharynx.
The spindle cell subtype of embryonal rhabdomyosarcoma has a spindled appearance and is frequently found in paratesticular sites.
Patients with embryonal rhabdomyosarcoma have intermediate outcome; the mesenchymal cells tend to differentiate into cross-striated muscle cells. Immunohistochemistry may demonstrate actin- or desmin-positive reactions. Ultrastructural studies exhibit evidence of myogenesis. The presence of cross-striations confirms the diagnosis.
Embryonal histology occurs in 60% of cases and is found most commonly in the orbit, head and neck, and genitourinary sites.
TABLE 48-3 International Classification of Rhabdomyosarcoma | ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||
The poor-prognosis group includes alveolar and undifferentiated sarcomas.
Approximately 20% of rhabdomyosarcomas are the alveolar subtype, most commonly found in adolescents with truncal, retroperitoneal, and extremity tumors. The projected outcome for this group is 54% at 5 years (24).
The pleomorphic type is extremely rare; many cases formerly classified as pleomorphic are currently considered to be malignant fibrous histiocytoma.
Cases previously classified as extraosseous Ewing’s sarcoma are now more appropriately included in the Ewing’s family of tumors and are managed as such.
A multidisciplinary approach using surgery, irradiation, and chemotherapy is critical in the management of rhabdomyosarcoma; the optimal sequence and specific application of each modality are under investigation.
In general, stage I tumors resected with an adequate margin require chemotherapy but not adjuvant irradiation (34). Patients with more advanced stages benefit from irradiation and multiagent chemotherapy.
Historically, orbital exenteration was used. However, this procedure should be reserved for salvage treatment and enucleation for the management of posttreatment ocular complications.Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree