Fig. 23.1
Clinical appearance of retinoblastoma. A large retinoblastoma before (left) and after (right) chemotherapy. Note the marked reduction in tumor size after chemotherapy. This view was obtained with a fundus camera that simulates the view with indirect ophthalmoscopy.
The differential diagnosis of retinoblastoma includes persistent hyperplastic primary vitreous, Coats’ disease (congenital retinal telangiectasis), ocular toxocariasis, endophthalmitis, retinopathy of prematurity and other conditions that can result in the appearance of a white pupil reflex.
The benign variant of retinoblastoma is referred to as retinoma or retinocytoma [14, 15]. These benign retinal tumors can be distinguished from retinoblastoma clinically by features such as calcification and surrounding chorioretinal atrophy, and histopathologically by the presence of well-differentiated retinal-like cells forming fleurettes and rosettes. The molecular pathogenesis of these lesions remains uncertain, but they can be found in the parents of children with sporadic heritable retinoblastoma and in patients with low-penetrance forms of retinoblastoma.
23.2.3 Histopathology
The cell of origin for retinoblastoma remains unclear but is most likely a retinal progenitor cell that has the capacity to differentiate along the lineage of photoreceptors and Mueller cells [16–18]. Retinoblastomas are composed of small round cells with large nuclei and minimal cytoplasm (Fig. 23.2). Tumor cells vary in the degree of differentiation, with Homer-Wright rosettes, Flexner-Wintersteiner rosettes, and fleurettes representing attempts at photoreceptor differentiation. However, unlike most cancers the degree of differentiation in retinoblastoma has no prognostic significance. Histopathologic features with indisputable prognostic significance include optic nerve and choroidal invasion [19].
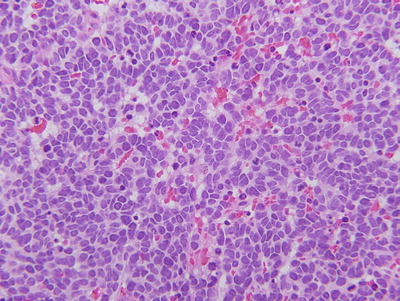
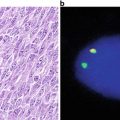
Fig. 23.2
Histopathologic appearance of retinoblastoma. Hematoxylin and eosin staining reveals small round blue cells with large nuclei and scant cytoplasm. This example is poorly differentiated.
23.2.4 Second Primary Tumors
Patients with heritable retinoblastoma have a systemic predisposition to cancer resulting from the germ line RB gene mutation present in most or all cells in the body. Multiple types of second primary cancers can be seen, including osteosarcomas, soft tissue sarcomas, melanomas, and epithelial cancers [8, 20]. The incidence of second primary tumors is roughly 25 % at 40 years after diagnosis of retinoblastoma [9]. If a patient receives radiation therapy, the risk of second malignancies increases significantly [21]. The finding of biallelic RB gene mutations in osteosarcomas and other second primary cancers lends support to the idea that the two-hit hypothesis of tumorigenesis also applies to these tumors [22].
23.2.5 Treatment and Prognosis
Over the past century there have been steady advances in the treatment of retinoblastoma [2]. Enucleation (eye removal) remains an important treatment option in certain cases (e.g., unilateral cases, very large tumors and invasion of the optic nerve or anterior segment). External beam radiotherapy (EBRT) was a mainstay of treatment for many years. However, the increased risk of second primary tumors and facial bone deformities associated with radiation has tempered enthusiasm for this treatment, particularly in patients younger than 12 months of age, who are at highest risk for these complications [23].
As an alternative to EBRT, several groups pioneered the use of chemotherapy in the 1990s [24, 25]. It was known from previous work several decades before that retinoblastoma responds to chemotherapy but frequently recurs due to the emergence of multidrug resistance. Therefore, the new approach combined chemotherapy with local consolidative techniques, such as laser and cryotherapy, to ablate the residual tumor masses after they had first been reduced in size by chemotherapy [24, 25]. Currently, the most frequently used agents are carboplatin, vincristine, and etoposide, given in 6–9 cycles. The survival rate in most developed countries is now greater than 95 %, but there are still many deficiencies that need to be addressed in the management of retinoblastoma. The risk of second primary tumors with current chemotherapy regimens remains unclear. Eyes with significant vitreous seeding generally do not do well with chemotherapy and often require radiation to salvage the eye. Intensive research efforts continue to identify new therapeutic options.
23.3 Molecular Perspectives
23.3.1 Molecular Genetics and Background
Retinoblastoma has played a pivotal role in cancer research that far outweighs its epidemiologic significance. Despite its rarity, retinoblastoma has provided unique and far-reaching insights into the genetics and molecular biology of cancer. The autosomal dominant inheritance of retinoblastoma has attracted the attention of cancer researchers for many years. The pivotal breakthrough occurred in 1971, when Alfred Knudson proposed his two-hit hypothesis that proposed that retinoblastoma results from the mutational inactivation of both alleles of a putative recessive oncogene [13]. The retinoblastoma locus was mapped to chromosome 13q through its linkage to esterase D, an enzyme that had previously been mapped to this region and which could be assayed for its enzymatic activity [26]. Esterase D activity was found to be about half the normal level in retinoblastoma patients with karyotypically visible deletions on chromosome 13q [27]. In another study, esterase D activity was 50 % of normal in healthy tissues of retinoblastoma patients but was undetectable in tumor tissue [28], providing further support for Knudson’s hypothesis . The recessive nature of the retinoblastoma locus was conclusively demonstrated by loss of heterozygosity in tumor tissue [29], a technique that subsequently has become a standard for studying tumor suppressor genes. Based on the proximity to the esterase D gene, several groups independently identified and characterized a nearby gene that proved to be the retinoblastoma gene [22, 30, 31]. Reintroduction of this gene into retinoblastoma cells suppressed the tumor phenotype, providing functional evidence for its role as a tumor suppressor [32].
23.3.2 The RB Gene
The retinoblastoma gene (RB) spans over 200 kilobases of DNA and contains 27 exons [33, 34]. Germ line mutations tend to cluster at CpG areas but are also spread throughout the gene with no mutational hot-spot [35, 36]. Germ line mutations can include deletions, frameshift and nonsense mutations, splicing mutations, and mutations in introns resulting in a truncated protein product [35]. The second hit is usually a nondisjunction or recombination event during mitosis, resulting in reduction to hemizygosity of the mutant allele [37]. Most RB mutations result in >90 % penetrance, but some mutations have been associated with low penetrance retinoblastoma. These low penetrance mutations can be divided into those that cause a reduction in the amount of normal Rb protein (usually promoter mutations) and those that result in a partially functional Rb protein (usually missense mutations) [38].
23.3.3 The Rb Protein
The retinoblastoma protein (Rb) is composed of 928 amino acids and has three functional domains: the N-terminus , the central pocket domain, and the C-terminus . The pocket, which is formed by the highly conserved A and B boxes, is required for binding to E2F, viral oncoproteins and chromatin remodeling proteins, and it is required for tumor suppression [39, 40]. Most RB mutations affect the pocket [35]. The C-terminus is also required for efficient binding to E2Fs, and it has binding sites for the oncoproteins MDM2 and c-abl [41–44]. The function of the N-terminus is less clear, and it does not appear to be required for tumor suppression [45].
A major function of Rb is to inhibit the G1–S phase transition of the cell cycle [46]. Protein activity is modulated during the cell cycle by phosphorylation, which can occur on 16 cyclin-dependent kinase Ser/Thr-Pro phosphoacceptor sites. Rb phosphorylation varies throughout the cell cycle, with hypophosphorylated Rb predominating in G0 and G1, and hyperphosphorylated Rb in S-phase and G2/M [47–49]. Rb usually becomes hypophosphorylated during differentiation and senescence, and it becomes hyperphosphorylated in cells reentering the cell cycle [50–52]. The Rb protein can also be modulated by viral oncoproteins, such as SV40 large T antigen, adenovirus E1a protein and herpes virus E7 protein, which bind and inhibit Rb, thereby stimulating cell cycle progression [53–55] (Fig. 23.4).
A major binding partner of Rb is the E2F family of transcription factors (E2Fs) [56]. Rb does not have a DNA binding domain but is brought to specific promoters through its interaction with E2Fs. E2F-responsive genes contain one or more E2F binding sites in their promoters, and these genes play an important role in cell cycle, apoptosis and other cellular functions [57]. In addition to a DNA binding domain, E2Fs 1–5 also have a transactivation domain by which they promote the transcription of specific genes. Rb inhibits transcription in part by binding and masking the E2F transactivation domain [58, 59]. In addition, Rb actively represses promoter and enhancer elements independently of its effect on the E2F transactivation domain [60, 61]. This so-called active repression is now thought to result from the recruitment by Rb of chromatin remodeling proteins that reconfigure local chromatin into a nonpermissive state for transcription [62]. These proteins include histone deacetylases, DNA methyltransferases, and polycomb complexes [63–68]. The functional significance of these two independent mechanisms of transcriptional repression by Rb remains controversial (Fig. 23.3). However, it seems clear that active repression by Rb is required for the full tumor suppressor activity of Rb, including its inhibition of the G1–S phase transition [69] (Fig. 23.4).
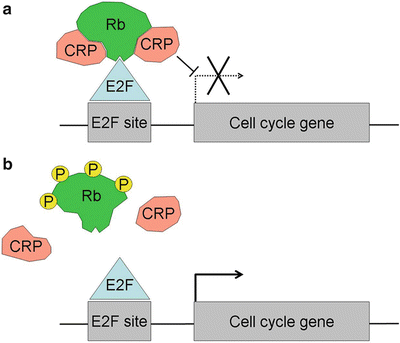
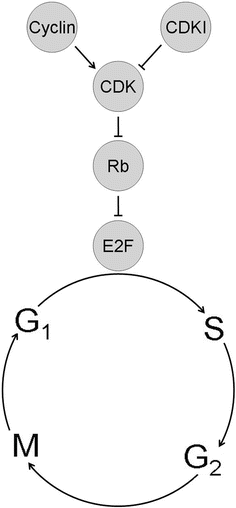
Fig. 23.3
The molecular mechanism of action of Rb. (a) When Rb is hypophosphorylated and active, it binds to E2F transcription factors that bind to E2F sites in the promoter regions of genes involved in cell cycle progression and apoptosis. Rb directly inhibits E2Fs by binding and masking the transactivation domain, and it also recruits chromatin remodeling proteins (CRPs) that alter local chromatin structure into a conformation that is not permissive for transcription. (b) When Rb is hyperphosphorylated, it does not bind efficiently to chromatin remodeling proteins or E2Fs, which allows E2Fs to transactivate cell cycle genes.
Fig. 23.4
The retinoblastoma tumor suppressor pathway. ) E2F transcription factors promote cell cycle progression in part by transactivating genes involved in DNA synthesis (S) phase of the cell cycle. Hypophosphorylated, active Rb inhibits cell cycle progression from G1 phase into S phase by directly inhibiting E2Fs and by actively repressing transcription of cell cycle genes. Rb can be inhibited by hyperphosphorylation catalyzed by cyclin dependent kinases (CDK). CDKs, in turn, require activation by cyclins, and they can be inhibited by CDK inhibitors (CDKIs) such as p16. Cancers tend to upregulate cyclins and CDKs and to downregulate CDKIs through genetic mutation or functional perturbation.
In addition to its role as a cell cycle inhibitor, Rb also inhibits apoptosis [46]. These seemingly opposing roles of Rb have not been satisfactorily reconciled. The widely accepted view of Rb as an on-off regulator of the G1–S transition that is completely inactivated every cell cycle does not explain why cells do not undergo apoptosis with every attempt at cell division. The most straightforward explanation is that Rb is not completely inactivated every cell cycle but that the degree of Rb phosphorylation and inactivation can be graded, depending on the cellular context. In support for this idea, Rb is phosphorylated in several hierarchical steps, and the phosphorylation events regulating the cell cycle can be differentiated from those regulating apoptosis [70, 71]. It seems likely that Rb is only partially phosphorylated and inhibited during normal cell division, and that this incomplete inactivation is sufficient to block its cell cycle inhibitory function and to allow cell cycle progression without triggering apoptosis [72]. This residual anti-apoptotic activity may persist unless Rb is more completely phosphorylated (or unless the RB gene is mutated), suggesting that Rb may serve as a buffer against apoptosis during normal cell division and a checkpoint for triggering apoptosis under abnormal stress conditions such as excessive mitogenic stimulation, DNA damage, and hypoxia in which apoptosis may be favored for survival of the organism.
23.3.4 The Rb Pathway
Although RB gene mutations are common in only a few types of cancer, such as retinoblastoma and small cell lung cancer, the RB pathway is functionally disrupted in virtually all human cancers [73, 74]. The RB pathway is composed of proteins that modulate Rb phosphorylation state. Rb is phosphorylated by cyclin-dependent kinases (CDKs) , which are activated by interaction with cyclins [74, 75]. Therefore, overactivity of cyclins or CDKs can lead to functional inactivation of Rb. For example, cyclin D1 is a bona fide oncogene that is overexpressed and functionally inhibits Rb in many cancer types [76, 77]. A constitutively active CDK4 mutant that maintains Rb in a phosphorylated state has been identified in some cancers [78]. On the other hand, CDKs are inhibited by proteins such as p16INK4a, which is a tumor suppressor that is mutationally inactivated in some cancers [79]. Interestingly, the cancers associated with complete Rb inactivation by genetic deletion (e.g., retinoblastoma and small cell lung cancer) have a different phenotype than those associated with partial Rb inhibition by loss of p16Ink4a or overexpression of cyclin D (e.g., melanoma, most solid tumors). The former tend to exhibit rapid proliferation, high apoptotic rates and sensitivity to chemotherapy, whereas the latter tend to have slow rates of proliferation and apoptosis and tend to be resistant to chemotherapy [80].
Related posts:




Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


