Retinoblastoma
Richard L. Hurwitz
Carol L. Shields
Jerry A. Shields
Patricia Chévez-Barrios
Dan S. Gombos
Mary Y. Hurwitz
Murali M. Chintagumpala
INTRODUCTION
Retinoblastoma, a malignant tumor of the embryonic neural retina, is the most common intraocular malignancy in children. Although usually not recognized at birth, retinoblastoma predom-inantly affects young children. The tumor has a variable growth rate, can originate from single or multiple foci in one or both eyes, and, in bilateral cases, may be manifest in one eye many months before it is evident in the other. Retinoblastoma is caused by a mutation in a gene that expresses a protein central to the control of the cell cycle and may occur sporadically or be inherited. Children with the hereditary type of retinoblastoma have a particular susceptibility to developing other malignant tumors. This disease serves as a model for understanding the genetics and heredity of child-hood cancer.
The term retinoblastoma was first adopted by the American Ophthalmological Society in 1926.1 The cellular origin of retinoblastoma had been a topic of debate since 1809, when, based only on gross pathological findings, the Scottish surgeon Wardrop first recognized that retinoblastoma is a discrete tumor arising from the retina.2,3 After this was published, other pathologists, including Robin and Langenbeck, confirmed the observations at a microscopic level. Virchow, however, thought that the cell of origin was glial and named it “glioma of the retina.” In the late 1800s, the term neuroepithelioma was proposed by Flexner and supported later by Wintersteiner because they believed that the tumor originated from the neuroepithelium and that the typical rosettes that now bear their names were attempts to form photoreceptors. In the early 1900s, Verhoeff concluded that the tumor was derived from undifferentiated embryonic retinal cells called retinoblasts and proposed the term retinoblastoma.1 Zimmerman proposed the term retinocytoma for the well-differentiated tumor that displays benign features.4 For this same tumor, Gallie et al.5 described the clinical features and proposed the term retinoma. The histopathological, ultrastructural, immunohistochemical and molecular characteristics of retinoblastoma support the concept that this tumor originates from a multipotent precursor cell. This cell could develop into almost any type of inner or outer retina cell including photoreceptors.6,7,8,9,10,11,12,13,14,15 Alternatively, investigations have supported the concept that a differentiated cell could be the precursor cell by de-differentiating into a malignant cell.16,17 That the benign retinoma is a precursor to the malignant retinoblastoma has also been proposed.18
EPIDEMIOLOGY
The Third National Cancer Survey predicts an average incidence of 11 new cases of retinoblastoma per million population less than 5 years of age, or 1 in 18,000 live births in the United States,19 and this rate has proven stable over time.20 Although data from developing countries are less complete, oncologists in Central and South America, the Middle East, and India generally feel that the incidence may be greater in these regions.21,22 A multicenter report from Mexico concluded that retinoblastoma is the second most frequent solid malignancy after CNS tumors in children.23 The estimated frequency of bilateral retinoblastoma ranges from 25% to 35% of reported cases.20,24 Thus, in the United States, an estimated 300 children per year develop retinoblastoma; of these 300, 60 to 90 cases are bilateral. There are no racial or gender predilections.
Retinoblastoma is often present at birth and is almost entirely restricted to early childhood. About 80% of cases are diagnosed before 3 to 4 years of age, with a median age at diagnosis of 2 years.20,21,24,25 The discovery of retinoblastoma beyond the age of 6 years is rare. Bilateral disease is diagnosed earlier than unilateral disease. Sporadic bilateral retinoblastoma has been associated with advanced parental age, but a recent analysis shows no increase in incidence of retinoblastoma with advancing maternal age.26
Multiple congenital anomalies associated with retinoblastoma have been reported in approximately 0.05% of US patients with retinoblastoma.27 The reported anomalies include congenital cardiovascular defects, cleft palate, infantile cortical hyperostosis, dentinogenesis imperfecta, familial congenital cataracts, and incontinentia pigmenti or Bloch-Sulzberger syndrome (an X-linked inherited disease that is lethal in males but affects females with pigmentary retinopathy, corneal opacities, cataracts, nystagmus, blue sclerae, myopia, pseudoglioma, dental abnormalities, abnormal skin pigmentation, and mental deficiency).25 An association with mental retardation has been suggested in children with the D-deletion syndrome; however, most patients with retinoblastoma have no intellectual impairment.
GENETICS
The majority of retinoblastomas appear sporadically; however, an inherited form of the disease has been documented28 and is transmitted with few exceptions as a typical Mendelian autosomal dominant trait with high but incomplete penetrance. Of all cases, about 60% are nonhereditary and unilateral, 15% are hereditary and unilateral, and 25% are hereditary and bilateral.29,30
A two-hit model has been proposed to explain the observations that familial cases are generally multifocal and bilateral, whereas sporadic cases typically present with unilateral unifocal disease at a later age.29,31 According to the model, as few as two stochastic mutational events are required for tumor initiation, the first of which can be inherited through the germ line (in heritable cases) or can occur somatically in individual retinal cells (in nonheritable cases). The second event occurs somatically in either case and leads to tumor formation from each doubly defective retinal cell.
The presence of a microscopically visible deletion in one of the chromosome 13 homologues in constitutional cells of a small number of retinoblastoma patients was the first evidence that supported an inherited mechanism for retinoblastoma development. 32,33,34,35 Although the deletions varied between families, each deletion minimally encompassed chromosome 13, band q14.36,37 This chromosomal locus contains the RB1 retinoblastoma gene. In the two-hit model, such deletions in the germ line could act as the first hit and confer the risk of tumor formation as an autosomal dominant trait. The increasing resolution of cytogenetic
technology and the development of DNA probes for loci in the immediate vicinity of the RB1 gene locus have allowed the detection of more subtle genomic rearrangements. These techniques can be used to identify people who carry nonpenetrant mutations in the retinoblastoma susceptibility locus.38
technology and the development of DNA probes for loci in the immediate vicinity of the RB1 gene locus have allowed the detection of more subtle genomic rearrangements. These techniques can be used to identify people who carry nonpenetrant mutations in the retinoblastoma susceptibility locus.38
Patients without a gross 13th chromosomal deletion but who have bilateral or familial retinoblastoma have submicroscopic mutations at the RB1 locus similar to mutations that have been found in the tumor cells of patients with nonhereditary retinoblastoma. The second step in tumorigenesis in both heritable and nonhereditary retinoblastoma involves somatic alteration of the normal allele at the RB1 locus in such a way that the mutant allele is unmasked. Thus, the first mutation in this process, although it may be inherited as an autosomal dominant trait in the child, is in fact a recessive defect in the individual retinal cell. Elimination of the chromosome containing the wild-type allele followed by reduplication of the remaining mutant chromosome may be one mechanism by which the affected RB1 locus becomes homozygous within the cell.39,40 Other mechanisms of mutagenesis including the generation of point mutations, deletions and silencing of the promoter by methylation are frequently encountered in the second allele. The result is that the potential tumor cell becomes functionally recessive for the mutant allele.
Although the unmasking of predisposing mutations at the RB1 locus occurs in mechanistically similar ways in sporadic and heritable retinoblastoma, only the latter carries the initial mutation in each cell. Patients with heritable disease also seem to be at greatly increased risk for the development of second primary tumors, particularly osteosarcoma.41 This high propensity is genetically determined by the predisposing RB1 mutation. The notion of a pathogenetic association between these two rare tumor types was tested by determining the constitutional and osteosarcoma genotypes at restriction fragment length polymorphism (RFLP) loci on chromosome 13. The data indicated that osteosarcomas arising in patients with retinoblastoma had become homozygous specifically around the chromosomal region carrying the RB1 locus. Furthermore, these same chromosomal mechanisms eliciting losses of constitutional heterozygosity were observed in sporadic osteosarcomas, suggesting a genetic similarity in pathogenetic causality.
These studies provided data useful for the molecular isolation of the RB1 gene.42 The 200-Kb RB1 locus contains 27 exons that are transcribed into a 4.7-Kb transcript in normal human and rat tissues including brain, kidney, ovary, spleen, liver, placenta, and retina.43 Although the number of different types of tumors that occur as a result of inherited mutations in the RB1 locus is small, the broad tissue expression and species conservation of this gene suggest a common and potentially important role in the growth or differentiation of many cell types.44,45,46,47 Introduction of the wild-type gene into retinoblastoma and osteosarcoma cell lines using recombinant retroviral vector transfer resulted in a partial reversal of the tumorigenic phenotype.48,49 Further characterization of the complete RB1 genomic sequence50 allowed a rigorous cataloging of the different mutations affecting the gene in retinoblastoma tumors. Over 200 disease-causing mutations have been identified in the retinoblastoma genes of patients.38,51,52,53,54,55
The examination of sporadic cases of bilateral retinoblastoma showed that disease frequently arises subsequent to a new germ line mutation in the paternal allele followed by somatic alteration or loss of the maternally derived wild-type allele.56,57 This finding suggests either that mutations in the RB1 locus occur more commonly during spermatogenesis or that the paternal chromosome in the early embryo is at a higher risk of mutation. Analyses of sporadic osteosarcomas also showed preferential mutation of the paternal allele.58
Investigations of RB1 gene alterations at both the DNA and the RNA levels cumulatively reveal a strong correlative relationship between the lack of RB1 gene product RB protein and the appearance of retinoblastoma tumors. In addition to osteosarcomas, other tumor types contain mutations involving the retinoblastoma gene. Molecular analyses of small cell lung carcinomas have revealed RB1 structural abnormalities in approximately 15% of cases.59 Loss of heterozygosity for chromosome 13 has been detected in about 25% of breast cancers and related breast cancer-derived cell lines.60,61 However, a more detailed analysis of the effects of chromosome 13 mutations in tumors has been compiled and clearly shows that not all tumors are either a direct or an indirect result of loss of heterozygosity of the RB1 locus.62 The cumulative data suggest that only subsets of tumors may share a common pathogenetic mechanism that results from unmasking mutations affecting the tumor suppressing function of the RB protein.
The RB protein is comprised of 928 amino acids and has an estimated molecular mass of 110 kDa. The expressed protein has been shown to be primarily localized in the cell nucleus.63 Posttranslational phosphorylation of the RB protein in quiescent cells overrides growth suppression and allows cell division to occur.64 The RB protein also has a role in the regulation of the cell cycle of actively dividing cells. The unphosphorylated RB protein (p110RB) has been shown to bind E2F1, a transcription factor and a cell cycle regulator during the G1 stage of the cell cycle. The RB/E2F1 complex masks the E2F1 transactivation domain and inhibits surrounding enhancer elements, thereby causing transcription of E2F1-regulated genes to cease. The RB protein accomplishes this by physically associating with a histone deacetylase (HDAC1). This recruitment of the deacetylase to the E2F1 regulating domain by RB allows deactylation of histone, thereby modulating the local structure of the chromatin.65,66 Phosphorylation of the RB protein at the G1/S boundary results in the release of these transcriptional factors, allowing them to become positive transcriptional elements. Additional cell cycle-specific kinases become activated and facilitate the progression of the cells through G2 and M. At the completion of the cell cycle, phosphatases dephosphorylate the RB protein, allowing the protein to again sequester E2F1 and form an inactive complex. Thus, positive and negative regulation of transcription and, therefore, cell proliferation are linked to the phosphorylation cycle of the RB protein. In tumors in which RB protein is mutated or absent, these intracellular transcriptional elements are dissociated and free to promote consistent and uncontrolled progression through the cell cycle. Such behavior results in unchecked cell proliferation consistent with a malignant phenotype.
The viral oncoproteins of polyomaviruses (SV40), adenoviruses (Ad-2 and Ad-5), and papillomaviruses (HPV-16) have also been shown to complex with the RB protein.67,68,69 Because one function of these viral oncoproteins appears to be the creation of a cellular environment that is permissive for DNA synthesis, one of their modes of action may involve sequestration of the antiproliferative unphosphorylated RB protein. Releasing the cell from its negative regulation by RB might allow the cell to enter the S phase and synthesize DNA. Taken together, the data support a model in which the unphosphorylated form of RB is the species active in growth suppression.
Mutations in the RB1 gene have been associated with greater than 95% of all retinoblastomas; however, there are a small number of tumors where RB1 mutations fail to be found. In at least some of these tumors, the MYCN gene is amplified and appears to be associated with the malignant phenotype.70
The understanding of the complex interrelationships of the RB proteins, mutations in the RB proteins that lead to disease, and the metabolic consequences of these events in the cell have led to the investigation of novel therapies to treat a variety of cancers including retinoblastoma.71,72
Genetic Counseling
Approximately 40% of patients with retinoblastoma have the inherited form of the disease. Since the inherited form of retinoblastoma is transmitted as an autosomal dominant trait with high but incomplete penetrance, there is a 45% chance that any given child of the patient will inherit the disease. In addition, although there is high penetrance of the retinoblastoma phenotype, the possibility exists that one of the patient’s siblings could also develop retinoblastoma due to germ line mosaicism and low penetrant alleles even if neither of the parents was affected by the disease (see Chapter 2). All children with a family history of retinoblastoma should be screened shortly after birth by a qualified ophthalmologist to permit early detection of the disease and to increase the chance of ocular and vision salvage. The increased familial risks support the need for expert genetic counseling.
To effectively counsel patients and families of retinoblastoma patients, the underlying cause of the disease must be determined. Patients who present with bilateral disease can be assumed to have a germ line mutation in the RB1 gene. Patients with unilateral disease at presentation may also have an underlying germ line mutation. If a mutation in the RB1 gene is detected in the tumor, somatic cells should also be screened. A mutation initially detected in the somatic cells is presumptive evidence of a mutation in the germ line. Genetic testing for the presence of this specific mutation in siblings or offspring should be pursued. These children can then be aggressively surveyed for the presence of emerging tumors. If genetic testing is not pursued, then tumor surveillance is recommended for all siblings of the affected patient. Current recommendations suggest examination at birth and every 4 months for up to 4 years of age. For children with unilateral disease, genetic screening for RB1 mutations can be offered to families at the time of enucleation. Testing for patients with unilateral disease requires a sample of tumor and peripheral blood.38 Testing for patients with bilateral disease requires only a blood sample. This topic is further discussed in Chapter 2.
CLINICAL PRESENTATION
Most cases of retinoblastoma in the United States are diagnosed while the tumor remains contained within the eye (intraocular) without local invasion or distant metastases. In developing countries, however, the diagnosis is frequently made only after an enlarged eye (buphthalmos) or gross orbital extension is apparent. These patients more commonly present with local invasion.
The signs and symptoms of an intraocular tumor depend on its size and position. The most common presenting sign is leukocoria of one or both eyes (Fig. 27.1). Leukocoria, white pupillary reflex instead of the normal red reflex, is manifest when the tumor is large or has caused a total retinal detachment leading to a retrolental mass visible through the pupil. In some cases, a small tumor centered in the macula (central portion of the retina) can produce leukocoria. If vitreous hemorrhage occurs because of bleeding from the retinoblastoma vessels, the pupil can appear to have a dark red reflex (hemocoria) or black reflex (nigrocoria) instead of the white reflex typically seen in retinoblastoma73,74 (Fig. 27.2). The second most common presenting sign is strabismus. Loss of central vision from a tumor in the macula can result in a disruption of fusional ability and cause the affected eye to drift.
Other ophthalmic features accompany some cases of retinoblastoma and may indicate the necessity for immediate enucleation. Heterochromia (different color for each iris) can present as an initial sign of retinoblastoma secondary to iris neovascularization. The diagnosis of retinoblastoma should be excluded in children that present with this condition.15 Rubeosis iridis (neovascularization of the surface of the iris) occurs in approximately 17% of patients with retinoblastoma and in more than 50% of patients with advanced retinoblastoma requiring enucleation15 (Fig. 27.2). Extensive necrosis of the tumor and liberated angiogenic factors may be responsible for this neovascularization of the iris.
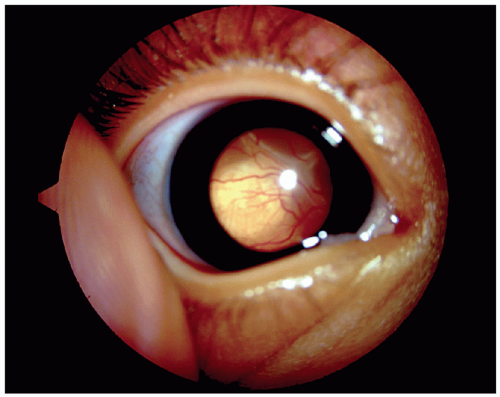 Figure 27.1 Photograph of an eye from a patient with retinoblastoma who presented with leukocoria. |
Spontaneous bleeding from rubeosis iridis can also cause hyphema (blood in the anterior chamber), and the potential diagnosis of retinoblastoma should be investigated in a child presenting with spontaneous hyphema without history of trauma.15,73,74 Glaucoma can be secondary to neovascularization of the anterior chamber angle and/or anterior synechia as a result of rubeosis iridis. Closed-angle glaucoma can also be secondary to mechanical obstruction of the anterior chamber angle by the iris and lens that has been pushed forward by a large intravitreal tumor. Most children with these presentations undergo enucleation.73,74 Anterior chamber seeding from endophytic tumors or diffuse infiltrating tumors may produce pseudohypopyon (cells in the anterior chamber)75 (Fig. 27.2). Intraocular tumors are not associated with pain unless secondary glaucoma or inflammation is present.
DIAGNOSIS
Most commonly, a parent or relative of an affected child notes an abnormality of the eye that prompts a pediatrician to look for leukocoria using an ophthalmoscope. The gross appearance of a creamy pink to snow white mass projecting into the vitreous (Fig. 27.1) may suggest retinoblastoma; however, associated findings of retinal detachment, vitreous hemorrhage, or opaque media often make inspection difficult. Pupillary dilation and examination with the patient under anesthesia are essential to fully evaluate the retina. Characteristically, the diagnosis is made by the ophthalmoscopic and ultrasonographic appearance, and pathologic confirmation is unnecessary. When the tumor is at an advanced stage, distinguishing vitreous seeding from multifocal tumors can be difficult; however, this distinction has important ramifications for the prognosis for the patient and for genetic counseling for the family. Earlier detection of the tumor would
benefit the patient both by decreasing the chance of a child presenting with metastatic disease and by increasing the chance of being able to salvage the affected eye. A suggestion has been made to include dilation of the pupil prior to examination at the first well-child visit. An additional benefit of screening would be the earlier detection and treatment of congenital and infantile cataracts. Whether routine screening would be practical is controversial because diseases such as retinoblastoma and congenital cataracts are rare (congenital cataracts affect approximately 1 in 2,000 live births) and because pediatricians may not be adequately trained to recognize these conditions.
benefit the patient both by decreasing the chance of a child presenting with metastatic disease and by increasing the chance of being able to salvage the affected eye. A suggestion has been made to include dilation of the pupil prior to examination at the first well-child visit. An additional benefit of screening would be the earlier detection and treatment of congenital and infantile cataracts. Whether routine screening would be practical is controversial because diseases such as retinoblastoma and congenital cataracts are rare (congenital cataracts affect approximately 1 in 2,000 live births) and because pediatricians may not be adequately trained to recognize these conditions.
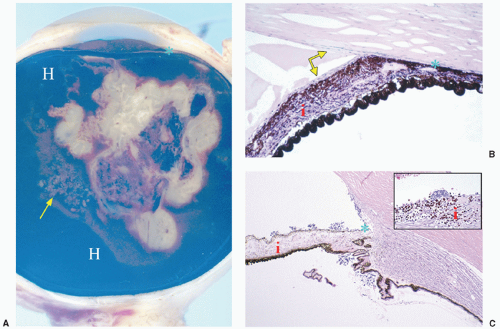 Figure 27.2 A: Gross photograph of an eye with retinoblastoma (white membranous tissue at center of the eye) with subretinal tumor seeds (arrow), and vitreous and subretinal hemorrhages (H). Neovascularization of the anterior chamber and partial closure of the anterior angle (*) are also present. B: Histologic picture of rubeosis iridis showing neovascularization (arrows) of the anterior portion of the iris (i) and focally on the endothelial surface of the cornea (arrow). Contraction of the neovascular membrane produces closure of the anterior chamber angle (*). Hematoxylin and eosin, original magnification 20×. C: Histologic picture of the anterior segment of an eye with retinoblastoma seeds on the surface of the iris (i) with focal rosette formation (inset). The anterior chamber angle (*) is opened. Hematoxylin and eosin, original magnification 20×. |
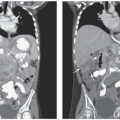
Ultrasonography, magnetic resonance imaging (MRI), and computed tomography (CT) of the orbit are the imaging studies most frequently used to confirm the diagnosis of retinoblastoma and to detect ectopic disease in the pineal gland. MRI of the orbit is a more useful technique to detect soft tissue tumor extension into the optic nerve and orbital coats. CT is better for detecting intraocular calcification that could provide confirmation of retinoblastoma (Fig. 27.3). However, in recent years, CT of children has fallen into disfavor due to the long-term risks for radiation-induced second malignancies.76
The pretreatment evaluation must be individualized for each patient. For patients who present with small tumors, careful scleral depressed examination in the office and later under anesthesia as well as office ultrasonography is necessary to make the diagnosis. A more extensive metastatic work-up is not necessary for these patients unless there is a question of optic nerve extension or extensive choroidal invasion. A lumbar puncture to obtain cerebral spinal fluid cytology and MRI or CT of the brain to rule out brain metastases can be performed for those patients. Because of the rarity of distant metastases in patients with retinoblastoma, a bone marrow examination or bone scan is usually not warranted unless the physician has suspicions of systemic involvement.
Differential Diagnosis
A number of benign conditions (pseudoretinoblastomas) can clinically simulate retinoblastoma and sometimes create considerable diagnostic difficulty for the ophthalmologist. Clinical definition is mandatory because the management of these entities differs considerably from the radical treatment of retinoblastoma. Early reports of the frequency of enucleations performed for suspected retinoblastomas when an alternative final pathological diagnosis is made varied from 30% to 16%, according to the degree of oncologic experience of, and the type of referrals received by, the group reporting the series.74 Most clinicians are now more familiar with pseudoretinoblastomas, and the frequency of erroneous enucleation is currently much lower.15
Other conditions that produce or simulate a mass in the vitreous or the retina and might be confused clinically are actually easily distinguished by a pathologist with the exception of medulloepithelioma.77 Approximately 60% of pseudoretinoblastomas include the differential diagnosis of three nonneoplastic entities: Toxocara canis endophthalmitis, persistent hyperplastic primary vitreous (PHPV) or Coats disease. All of these entities
might present with retinal detachment and may have retrolenticular fibrosis. Toxocara canis endophthalmitis is caused by the larvae of the nematode Toxocara canis and presents almost always in children, although never at birth. Clinical history and serology are important for the diagnosis. Usually there are no signs of ocular inflammation as the live larvae do not elicit an inflammatory response. Dead larvae elicit the formation of a localized eosinophilic abscess surrounding the microorganism. Condensed vitreous with gliosis and fibrosis may be present at the site of infection. Because these organisms are very small and degenerate, histological confirmation is difficult.
might present with retinal detachment and may have retrolenticular fibrosis. Toxocara canis endophthalmitis is caused by the larvae of the nematode Toxocara canis and presents almost always in children, although never at birth. Clinical history and serology are important for the diagnosis. Usually there are no signs of ocular inflammation as the live larvae do not elicit an inflammatory response. Dead larvae elicit the formation of a localized eosinophilic abscess surrounding the microorganism. Condensed vitreous with gliosis and fibrosis may be present at the site of infection. Because these organisms are very small and degenerate, histological confirmation is difficult.
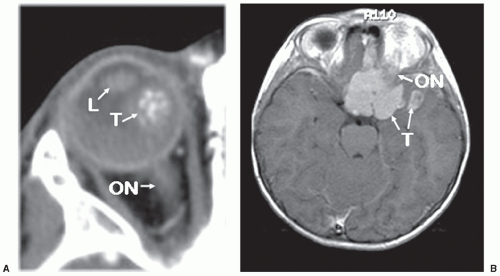 Figure 27.3 Diagnostic imaging of children with retinoblastoma. A: CT scan of the orbit. Axial view showing a partially calcified mass (T) consistent with retinoblastoma, a normal lens (L), and normal optic nerve (ON). The globe is intact and shows no evidence of extraocular invasion by tumor. B: MRI of the orbits and brain. Contrast-enhanced coronal T1-weighted image showing parasellar and left middle fossa spread of retinoblastoma (T) with extension along the sylvian fissure. The eye on the left is normal. The orbit on the right contains a prosthesis. |
PHPV (or Persistent Fetal Vasculature [PFV]) is a congenital anomaly of the primary vitreous where embryonal vessels do not regress and may pull the retina, resulting in an anterior retinal detachment. A posterior subcapsular cataract forms if the posterior capsule of the lens is ruptured by the traction of the vessels and fibrous membrane. Some cases of PHPV are associated with a wide band to the optic nerve and with retinal dysplasia. There are rare cases reported when PHPV has been associated with retinoblastoma.78
In contrast to toxocariasis and PHPV, Coats disease lacks the fibrosis and vascularization of vitreous. Coats disease is characterized by peripheral retinal vascular telangiectasia. These abnormal vessels leak and create an exudative retinal detachment rich in lipids with subretinal foamy macrophages and cholesterol clefts. Toxocariasis and PHPV simulate endophytic retinoblastoma, and Coats disease mimics the exophytic type (see “Pathology”). Ultrasound, CT scans, MRI, and other ancillary imaging technology have greatly helped in the differential diagnosis of these lesions.79
The Use of Cytology in the Diagnosis of Retinoblastoma
Retinoblastoma is one of the only human tumors radically treated without tissue biopsy confirmation. The clinical presentation and the ancillary radiologic and ultrasonic findings are typical for retinoblastoma in the majority of patients. Usually, the correct diagnosis does not represent a diagnostic dilemma for the experienced pediatric/oncologic ophthalmologist. The resistance to biopsy confirmation of the tumor arises from the dramatic difference between survivals for patients with contained intraocular tumors versus those with extraocular seeding of the tumor. The bias against biopsy is also aggravated by the reports of cases where the tumor was misdiagnosed as uveitis or obscured by cataract and patients developed orbital extensions of retinoblastoma after vitrectomy. Aggressive postvitrectomy therapy has resulted in prevention of metastasis in most patients with unsuspected retinoblastoma.80
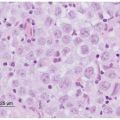
Recently, the development of more refined techniques of fine-needle aspiration biopsy (FNAB) and the increased knowledge of the biological behavior of retinoblastoma have allowed some patients to benefit from pretreatment biopsy.81,82 The technique of FNAB for retinoblastoma involves a transcorneal approach rather than the typical trans pars plana approach. The transcorneal technique is more difficult to perform; it is, however, safer for the patient because the needle penetrates several tissue layers before it enters the tumor, reducing the risk for extraocular spread. The 30-gauge needle passes through the peripheral cornea, anterior chamber, peripheral iris, lens zonules (avoiding puncture of the lens), vitreous, and penetrates the tumor (Fig. 27.4). The cytological findings are small- to medium-size basophilic cells with scanty cytoplasm that tend to group together (rosette-like). Mitoses may be easy to find, and necrosis is frequently encountered (Fig. 27.4). Similar features can be seen in cytologic CSF specimens in children with intracranial metastases (Fig. 27.4). There have been no reports of extraocular tumor spread through the needle tract after transcorneal FNAB, but FNAB is recommended only in selected cases where the diagnosis is ambiguous and when adequate steps are taken to prevent extraocular seeding of tumor cells.83 In one series of FNAB of pediatric intraocular tumors, the overall accuracy of FNAB was
95%, and the accuracy of cytologic interpretation was 100%. FNAB is, therefore, a reliable and accurate diagnostic tool for the assessment of selected pediatric ophthalmic diseases when the diagnosis is in question.84
95%, and the accuracy of cytologic interpretation was 100%. FNAB is, therefore, a reliable and accurate diagnostic tool for the assessment of selected pediatric ophthalmic diseases when the diagnosis is in question.84
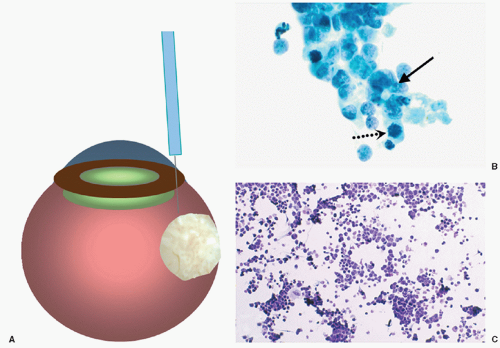 Figure 27.4 A: Drawing of an eye with retinoblastoma indicating the entrance of the needle through the peripheral cornea, peripheral iris, then between the ciliary body and the lens into the tumor. The technique avoids vascularized conjunctiva of the limbus and the orbit, sclera and pars plana preventing possible spreading of tumor cells via the needle tract. B: Cytologic preparation of a retinoblastoma showing cohesive groups of neoplastic cells with high nuclear:cytoplasmic ratio, increased mitotic activity (dotted arrow), and focal rosette formation (solid arrow). Papanicolaou stain, original magnification 100×. C: Cytologic preparation of cerebrospinal fluid in a patient with retinoblastoma metastatic to the brain. Notice the cohesive groups of neoplastic cells with high nuclear:cytoplasmic ratio. Hematoxylin and eosin stain. Original magnification 20×. |
PATHOLOGY
Guidelines for Processing Eyes with Retinoblastoma
To obtain an adequate specimen for evaluation of tumor characteristics, histopathologic risk features (described later), and genetic evaluation, it is necessary to process the freshly enucleated eye and obtain tumor without excessive contamination of the intraocular structures. Sections of the eye should include the structures to evaluate (choroid, tumor, all levels of the optic nerve, and anterior chamber structures).85 In summary, there are three recommended techniques to obtain fresh tumor: (a) creation of a chorioscleral window with a blade (Fig. 27.5), (b) creation of a chorioscleral window with a corneal trephine, or (c) aspiration of tumor with a wide-bore syringe. Any of these techniques should be performed immediately after enucleation with awareness of the anatomic orientation and taking care to avoid collapsing the eye or excessively contaminating ocular structures with tumor. Before opening the eye, a cross section of the optic nerve should be obtained and submitted for histopathologic examination to avoid contamination by tumor. Tumor for genetic analysis is then retrieved and immediately frozen at -70°C to -80°C in cryoresistant tubes (Fig. 27.5). The eye is then placed in 10% formalin and, after adequate fixation, the eye is grossed to obtain a central section including the optic nerve, tumor, and pupil (P.O. pupil-optic nerve section), and two calottes (remainder of eye). The calottes should be further sectioned into anterior and posterior segments and submitted on edge for histopathologic examination of the choroid. In total, there should be 4 blocks: one with the optic nerve surgical margin cross section, one with the PO section, and one for each calotte in segments (Fig. 27.6). Several levels of each block should be reviewed for diagnosis.
Gross Features
Primary retinoblastomas originate in the sensory retina and occupy the retina and vitreous cavity. Retinoblastoma tumors are usually white-gray with a chalky appearance and a soft, friable consistency. Bright white speckles corresponding to calcifications present throughout the tumor. The gross features of retinoblastoma depend on the growth pattern of the tumor.73,86 Some of these patterns correlate with clinical presentations and differences in biological behavior, especially as they relate to intraocular and extraocular types of tumor spread.
The endophytic growth pattern is represented by tumors arising from the retina and growing into the vitreous cavity (Fig. 27.7). These tumors tend to entirely fill the cavity and produce floating
tumor spheres called vitreous seeds. A tumor left untreated eventually invades the anterior portion of the eye, reaching the aqueous venous channels and the conjunctiva. From there, the tumor can permeate the lymphatic vessels and metastasize to regional lymph nodes.86,87,88
tumor spheres called vitreous seeds. A tumor left untreated eventually invades the anterior portion of the eye, reaching the aqueous venous channels and the conjunctiva. From there, the tumor can permeate the lymphatic vessels and metastasize to regional lymph nodes.86,87,88
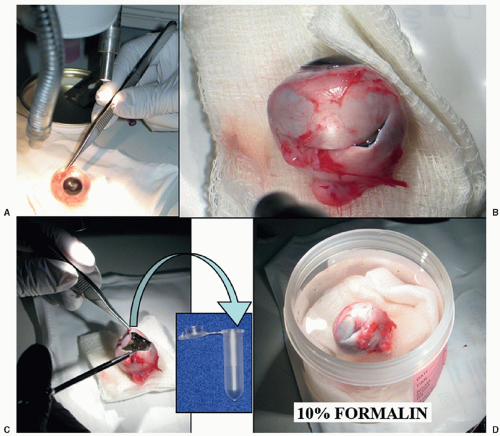 Figure 27.5 Handling of the enucleated eye with retinoblastoma for tumor retrieval. A: Preferably under a stereoscopic microscope, the eye is examined to select the area in which most of the tumor is present. This is achieved by retroillumination to identify the area of the tumor by the denser shadow. Before sectioning into the eye, one must obtain the cross section of the optic nerve margin for histologic examination. The scleral window should be created perpendicular to this site to obtain tumor at the edge of the larger mass. B: The opening of the eye is made with a sharp blade or by using a corneal threphine (not shown here), to obtain a scleral window. C: Under the miscroscope, select the areas of least necrosis and calcifications without disturbing intraocular structures such as retina and optic nerve head. Retrieve tumor and place in cryoresistant tubes to immediately freeze and store until needed for genetic studies or research. D: Place the eye in 10% formalin, gently reforming the round shape of the eye, and fix for at least 24 hours. |
Exophytic tumors grow from the retina into the subretinal space and often cause serous detachments of the retina (Fig. 27.7). These tumors may invade the choroid through Bruch’s membrane.86,89 Mixed endophytic and exophytic tumor growth is the most common pattern encountered15,86 (Fig. 27.7).
Diffuse infiltrating retinoblastoma is the least common tumor growth pattern, but is the most diagnostically challenging because there is no predominant mass (Fig. 27.7). This presentation of retinoblastoma is seen in children with an average age of 6 years. The tumor cells grow throughout the retina, while single cells and vitreous seeds invade the anterior portions of the retina, the ciliary body, and, eventually, the anterior chamber. Clinically, this type of tumor resembles an inflammatory process presenting as pseudohypopyon mimicking inflammatory cell accumulation (hypopyon) in the anterior chamber and vitreous seeds simulating the inflammatory cellular reaction seen in uveitis. Because this type of retinoblastoma resembles an inflammatory process, the diagnosis is often delayed until cytological examination of the aqueous humor or, in rare cases, of the vitreous. Almost all reported cases have a unilateral, sporadic presentation without family history. Although the diagnosis is difficult, children with diffuse infiltrating retinoblastoma have a good prognosis after enucleation.88,90 Any child, regardless of age, who presents with signs of endophthalmitis should be considered to have diffuse infiltrating retinoblastoma until proven otherwise.
Another unusual presentation is the extensively necrotic retinoblastoma that presents as severe inflammatory reaction with massive necrosis of the tumor (more than 95%) and with necrosis of the intraocular structures. If the eye is left untreated, this may be followed by phthisis bulbi (complete atrophy of the eye).91 If the eye is enucleated at the time of acute necrosis, the gross findings are those of massive tumor necrosis, intraocular tissue necrosis with dispersion of pigment, and edema of the conjunctiva
(Fig 27.8). Extensively necrotic retinoblastoma is associated with a statistically significant increase in histopathologic risk factors such as choroidal invasion and optic nerve invasion.92 Through unknown mechanisms, complete spontaneous tumor regression occurs more commonly in retinoblastoma than in any other malignant tumor. In most of these cases, complete occlusion of the central retinal artery is found; however, it is not known whether this is a primary event or the result of tumor necrosis. If the eye is examined after complete atrophy has occurred, the findings are those of a small shrunken eye with mostly necrotic calcified tumor and a disorganized retina.
(Fig 27.8). Extensively necrotic retinoblastoma is associated with a statistically significant increase in histopathologic risk factors such as choroidal invasion and optic nerve invasion.92 Through unknown mechanisms, complete spontaneous tumor regression occurs more commonly in retinoblastoma than in any other malignant tumor. In most of these cases, complete occlusion of the central retinal artery is found; however, it is not known whether this is a primary event or the result of tumor necrosis. If the eye is examined after complete atrophy has occurred, the findings are those of a small shrunken eye with mostly necrotic calcified tumor and a disorganized retina.
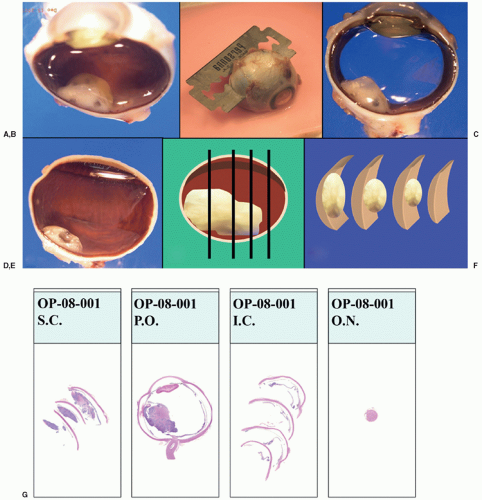 Figure 27.6 Grossing of the eye with retinoblastoma. A: After adequate fixation, remove the calotte where the window to obtain fresh tumor was created by cutting nearer to the optic nerve (without transecting it) and to avoid the opened area of the sclera. B: Cut and remove the opposite calotte to obtain the central pupil optic nerve section (P.O.) C: Central P.O. section showing anterior chamber, pupil, lens, tumor, and optic nerve in one section. D: Calotte with some tumor. E: Schematic representation of the anterior-posterior sections of the calottes. F: Schematic representation of the sections already cut from the calottes to submit for processing into paraffin blocks and slides. G: At the end of the process, there should be four blocks that give rise to slides with sections of the calottes (2), the PO sections (1), and the cross section of the optic nerve margin (1).
Related posts:Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

|