Chapter 69 Retinoblastoma
Retinoblastoma is the most frequent neoplasm of the eye in childhood, representing 2.5% to 4% of all pediatric cancers. The average age-adjusted incidence rate of retinoblastoma in the United States and Europe is 2 to 5 per million children (approximately 1 in 14,000 to 18,000 live births).1,2 Retinoblastoma is a cancer of the very young; two thirds of all cases are diagnosed before 2 years of age, and 95% of cases are diagnosed before 5 years of age.1
Etiology and Epidemiology
The incidence of retinoblastoma is higher (6 to 10 cases per million children) in Africa, India, and children of Native American descent in North America. The increased incidence occurs primarily in unilateral cases. In industrialized countries, an increased incidence of retinoblastoma is associated with poverty and low levels of maternal education, that suggests environmental factors in its etiology.3,4 Recent studies have suggested a role for human papillomaviruses in the pathogenesis of retinoblastoma. The viral oncoprotein E7 of high-risk human papillomavirus types has been shown to bind to and inactivate the RB1 gene product (pRB), acting functionally to create the biallelic loss of RB1.5,6 High-risk human papillomavirus sequences have been detected in 28% to 36% of tumors.6
Biology
In 1971, based on the mathematical analysis of the age at presentation of bilateral (hereditary) and unilateral (mostly nonhereditary) cases of retinoblastoma, Knudson7 proposed the “two-hit hypothesis,” in which two mutational events in a developing retinal cell lead to the development of retinoblastoma. This hypothesis was subsequently extended to suggest that the two events could be mutations of both alleles of the RB1 gene, located on chromosome 13q14 and identified in 1986.8,9 Its product, pRb, is required for the transition of the cell through the G1 phase of mitosis. pRb appears to function as a tumor suppressor at least in part by inhibiting cell cycle progression past the G1-S restriction point; on entry to the S phase, cells become irreversibly committed to division. pRb is the major gatekeeper to control this critical point in growth regulation; lack of pRb activity removes the pRb constraint on cell cycle control, resulting in deregulated cell proliferation.
The RB1 gene is a large gene, containing 27 exons over about 200 kilobases of DNA, and mutations have been described in almost every exon.10 New germline mutations have an overwhelming preference for the paternal allele. Penetrance of the trait is greater than 90%.11
In both hereditary and nonhereditary retinoblastoma the second tumorigenic event is usually chromosomal, often as a result of mitotic recombination errors.12 This second hit occurs at a much higher frequency than the first hit, and it is more sensitive to environmental factors such as ionizing radiation, perhaps explaining the increased risk of radiation-induced malignancies in survivors of retinoblastoma.
Prevention, Early Detection, and Genetic Counseling
The successful management of retinoblastoma depends on the ability to detect the disease while it is still intraocular. Disease stage correlates with delay in diagnosis.14 In developing countries, late diagnosis is associated with orbital and metastatic disease.15 Eye assessment is encouraged in all neonates and at subsequent health visits. Mass screening is being considered, especially where the tumor is common in areas of South America and Asia.
Retinoblastoma is a unique neoplasm in that the genetic form imparts a predisposition to developing tumor in an autosomal dominant fashion with almost complete penetrance (85% to 95%).16 The majority of such children acquire the first mutation as a new germline mutation, with only 15% to 25% having a positive family history. However, some families display an inheritance pattern characterized by reduced penetrance and expressivity. Also, the RB1 gene mutation can occur at a late stage of embryogenesis, resulting in a variable expression depending on the tissue, causing mosaicisms in 10% to 15% of the patients or their progenitors.17 Recognizing the inheritance pattern and the existence of mosaicisms, the following familial risk estimates can be made16:
Genetic counseling is of the utmost importance to assist parents in understanding the genetic consequences of each form of retinoblastoma and to estimate the risk in relatives. With the refinement in methods of mutational analysis over the past decade, detection rates have increased to more than 90%.10
Pathology and Pathways of Spread
Macroscopically, retinoblastoma is soft and friable, tending to outgrow its blood supply with resultant necrosis and calcification. Dissemination within the vitreous and retina in the form of small, white nodules (seeds) is common, sometimes making it difficult to distinguish a multicentric primary tumor from a disseminated tumor.18 The microscopic appearance depends on the degree of differentiation. Undifferentiated retinoblastoma is composed of small, round, densely packed cells with hypochromatic nuclei and scant cytoplasm. Several degrees of photoreceptor differentiation have been described, characterized by distinctive cellular arrangements. Flexner-Wintersteiner rosettes (clusters of low columnar cells arranged around central lumens bounded by eosinophilic membranes analogous to the external membrane of the normal retina) are specific for retinoblastoma and are noted in 70% of tumors. Fleurettes (composed of larger cells with abundant eosinophilic cytoplasm arranged in a distinctive fleur-de-lis pattern) are less frequent; cells exhibit ultrastructural characteristics of photoreceptor differentiation. Especially well-differentiated tumors composed almost entirely of fleurettes have been called retinomas or retinocytomas. Ultrastructurally, retinoblastoma cells demonstrate photoreceptor differentiation with the presence of the 9-0 microtubule doublet pattern, abundant cytoplasmic microtubules, synaptic ribbons, and neurosecretory granules.
Clinical Manifestations, Patient Evaluation, and Staging
Retinoblastoma is a tumor of young children. Bilateral retinoblastoma presents at a younger age (usually before 1 year of age) than patients with unilateral disease (typically at age 1 to 3 years).16,18,19 Half the cases of retinoblastoma diagnosed during the first year are bilateral, compared with fewer than 10% when diagnosed after 1 year of age.
Although most patients with bilateral retinoblastoma carry a germline mutation of the RB1 gene, only 5% carry a deletion involving the 13q14 locus large enough to be detected by karyotyping. In those cases, retinoblastoma is part of a more complex syndrome resulting from the loss of additional genetic material. Patients with the 13q− syndrome are characterized by typical facial dysmorphic features, subtle skeletal abnormalities, and varying degrees of mental retardation and motor impairment.20 The severity of deficits correlate with the size of the deletion; normal psychomotor development may be seen in those patients in whom the deletion is restricted to the 13q14 band.
Trilateral retinoblastoma refers to the association of bilateral retinoblastoma with a typically asynchronous brain tumor.21 The majority of tumors are pineal region primitive neuroectodermal tumors (PNETs, pineoblastomas); 20% to 25% of the tumors are suprasellar or parasellar. The PNETs exhibit varying degrees of neuronal or photoreceptor differentiation, suggesting an origin from the germinal layer of primitive cells. Trilateral disease occurs in 3% to 9% of patients with the genetic form of retinoblastoma and appears to be more common in familial cases. The prognosis until recently has been almost uniformly fatal. The median age at diagnosis of trilateral retinoblastoma is 23 to 48 months; the interval between initial diagnosis of bilateral retinoblastoma and the diagnosis of the brain tumor is usually more than 20 months.22 In recent years, with the more widespread use of chemoreductive therapy and less use of radiation therapy (RT) for patients with bilateral retinoblastoma, the incidence of trilateral retinoblastoma has decreased dramatically.23 Approximately 5% of patients with bilateral disease develop pineal cysts, which appear to be a forme fruste of trilateral retinoblastoma.24
The diagnosis of intraocular retinoblastoma is usually made without pathologic confirmation. An examination under anesthesia with a maximally dilated pupil and scleral indentation is required to examine the entire retina (Fig. 69-1). Endophytic tumors are those that grow inward to the vitreous cavity and may seed the vitreous cavity. Exophytic retinoblastoma grows into the subretinal space, causing progressive retinal detachment and subretinal seeding. A very detailed documentation of the number, location, and size of tumors, of the presence of retinal detachment and subretinal fluid, and of the presence of vitreous and subretinal seeds must be performed. Wide-angle real-time retinal imaging systems such as RetCam (Clarity Medical Systems, Pleasanton, Calif.) provide a 130-degree field of view and digital recording, thus facilitating diagnosis and follow-up.
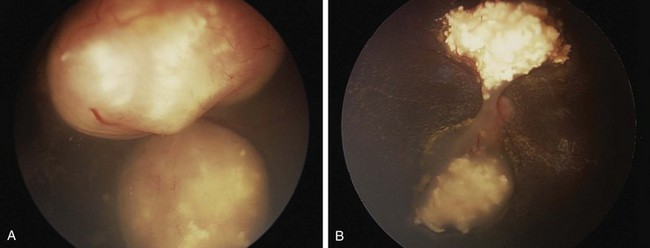
Imaging studies that aid in the diagnosis include bidimensional ultrasonography, computed tomography, and magnetic resonance imaging (MRI). Imaging is particularly important to evaluate extraocular extension and to differentiate retinoblastoma from other causes of leukocoria. Evaluation for the presence of metastatic disease also needs to be considered in a subgroup of patients. Metastatic disease occurs in 10% to 15% of patients, usually in association with advanced intraocular features, such as deep choroidal and scleral invasion, or involvement of the iris or ciliary body or of the optic nerve beyond the lamina cribrosa.13 In such cases, additional staging should include bone scintigraphy, bone marrow aspiration and biopsy, and lumbar puncture.
The Reese-Ellsworth grouping system has been the standard for intraocular disease. This system was designed to predict the outcome after external beam radiation therapy (EBRT). It divides eyes into five groups on the basis of the size, location, and number of lesions and on the presence of vitreous seeding25 (Table 69-1). The changed paradigm for “conservative” chemoreduction strategies for intraocular retinoblastoma has made the Reese-Ellsworth system less predictive of ocular preservation. A new staging system (i.e., the International Classification of Retinoblastoma) has been developed to provide a simpler, more user-friendly classification applicable to current therapies. The new system is based on the extent of tumor seeding within the vitreous cavity and subretinal space and seems to be a better predictor of treatment success26 (Table 69-2).
TABLE 69-1 Reese-Ellsworth Grouping for Suitability for Treatment of Retinoblastoma by Radiation Therapy
| Group | Description |
|---|---|
| I (Very Favorable) | |
| Ia | Solitary tumor smaller than 4 dd at or behind the equator |
| Ib | Multiple tumors, none larger than 4 dd, all at or behind equator |
| II (Favorable) | |
| IIa | Solitary tumor 4-10 dd, at or behind equator |
| IIb | Multiple tumors 4-10 dd, at or behind equator |
| III (Doubtful) | |
| IIIa | Any lesion anterior to equator |
| IIIb | Solitary tumor larger than 10 dd behind equator |
| IV (Unfavorable) | |
| IVa | Multiple tumors, some larger than 10 dd |
| IVb | Any lesion extending anteriorly to the ora serrata |
| V (Very Unfavorable) | |
| Va | Massive tumors involving more than half the retina |
| Vb | Vitreous seeding |
dd, disc diameter (1.5 mm).
From Reese AB, Ellsworth RM: The evaluation and current concept of retinoblastoma therapy, Trans Am Acad Ophthalmol Otolaryngol 67:164-172, 1963.
TABLE 69-2 International Classification of Retinoblastoma
| Group A |
| Group B |
| Group C |
| Group D |
| Group E |
Modified from Shields CL, Mashayekhi A, Au AK, et al: The International Classification of Retinoblastoma predicts chemoreduction success, Ophthalmology 113:2276-2280, 2006.
For patients undergoing enucleation, pathologic staging incorporates features known to influence outcome, guiding the therapeutic approach; such factors include choroidal and scleral involvement, optic nerve extension, and presence of metastatic disease. A third proposed staging system developed by an international consortium of ophthalmologists and pediatric oncologists incorporates the most important elements of the older systems27 (Table 69-3). Growth and invasion occur as a sequence of events, and extraretinal extension occurs only after the tumor has reached large intraocular dimensions. As part of this process, retinoblastoma extends into the ocular coats (choroids and sclera), the optic nerve, and the anterior segment. Extraocular disease is the next step in this progression; locoregional dissemination occurs by direct extension through the sclera into the orbital contents and preauricular lymph nodes. Extraorbital disease manifests as intracranial tumor, leptomeningeal dissemination, or hematogenous metastasis.
TABLE 69-3 International Retinoblastoma Staging System, Proposed
| Stage | Treatment |
|---|---|
| 0 | Patients treated conservatively |
| I | Eye enucleated, completely resected histologically |
| II | Eye enucleated, microscopic residual tumor |
| III | Regional extension |
| IIIa | Overt orbital disease |
| IIIb | Preauricular or cervical lymph node extension |
| IV | Metastatic disease |
| IVa | Hematogenous metastasis (without CNS involvement) |
| IVb | CNS extension (with or without any other site of regional or metastatic disease) |
From Chantada G, Doz F, Antonelli CB, et al: A proposal for an international retinoblastoma staging system, Pediatr Blood Cancer 47:801-805, 2006.
Primary Therapy
Principles of Treatment
Treatment of retinoblastoma aims to save life and preserve useful vision, with attention to late functional and carcinogenic effects. Contemporary care requires coordination among the patient’s ophthalmologist, pediatric oncologist, and radiation oncologist.28
Surgery
Enucleation is indicated for large tumors filling the vitreous when there is little or no likelihood of restoring vision and when tumor is present in the anterior chamber or associated with neovascular glaucoma. The eye must be removed intact, without seeding tumor cells into the orbit.29 For optimal staging, a long segment (10 to 15 mm) of the optic nerve is removed with the globe. An orbital implant is usually fitted during the same procedure, and the extraocular muscles are attached to it. A ceramic false eye is later fitted in the orbital socket. Orbital exenteration is very seldom indicated. For patients presenting with orbital (extraocular) disease, judicious use of chemotherapy, surgery, and RT provides tumor control and often orbital exenteration can be avoided.
Focal Surgical Therapies
Focal treatment is used for small tumors (<3-6 mm), classically in patients with bilateral disease and in combination with chemotherapy. Focal therapies are central to ongoing studies utilizing conservative therapy (chemotherapy and focal treatment) to avoid enucleation and EBRT in unilateral intraocular disease. Photocoagulation with the argon laser is used for the treatment of tumors situated at or posterior to the equator of the eye and for the treatment of retinal neovascularization occasionally seen after RT.30 This technique is limited to tumors measuring no greater than 4.5 mm at its base and no greater than 2.5 mm in height. The treatment aims to obliterate blood supply to the tumor. Two or three monthly sessions are usually required, and local tumor control is achieved in approximately 70% of cases. Cryotherapy is used for the treatment of small equatorial and peripheral lesions that measure no more than 3.5 mm at the base and no more than 2 mm in height.31 One or two monthly sessions of triple freeze and thaw are performed; the resultant tumor control is usually excellent. A more recently introduced focal therapy is transpupillary thermotherapy, which applies focused heat at sub-photocoagulation levels, usually with a diode laser.32 In thermotherapy, the goal is to deliver a temperature of 42° C to 60° C for 5 to 20 minutes to the tumor, relatively sparing retinal vessels from photocoagulation. The use of focal treatments is especially important in conjunction with chemotherapy, when both treatment modalities appear to have synergistic effects. Sequential thermotherapy and carboplatin enhances the antitumor effect by increasing the platinum-DNA adducts; thus, thermochemotherapy is becoming an important component of therapy for intraocular retinoblastoma.33 In general, local control rates of 70% to 80% can be achieved. Complications of focal treatments include transient serous retinal detachment, retinal traction and tears, and localized fibrosis.
Chemotherapy
Chemotherapy is indicated in patients with extraocular disease, in the subgroup of patients with intraocular disease with high-risk histologic features, and in patients with bilateral disease in conjunction with aggressive focal therapies. In patients with early-stage unilateral disease, chemotherapy is central to conservation therapy when combined with focal therapy. Agents effective in the treatment of retinoblastoma include platinum compounds, etoposide, cyclophosphamide, doxorubicin, vincristine, and ifosfamide.28,34
Radiation Therapy
The spectrum of radiation techniques includes local brachytherapy plaques, focal external beam irradiation (intensity-modulated radiation therapy [IMRT] or stereotactic photon RT, or proton beam), full ocular EBRT (with the same technical options as just mentioned, recent reports documenting techniques to limit dose to the bony orbit and contralateral eye), and orbital irradiation.35–47 Recognizing the balance of potential secondary carcinogenesis (particularly in bilateral or hereditary retinoblastoma) and orbital growth changes, while noting the long documented efficacy of RT in this disease, challenges the radiation oncologist as to indications and timing for management, optimal treatment planning and delivery, and requisite follow-up.48–50
Intraocular Retinoblastoma
Unilateral Retinoblastoma
For unilateral intraocular presentations, enucleation is curative for 85% to 90% of children. Outcome after enucleation is excellent, with good functional results and minimal long-term effects.48 In view of the apparent success in treating bilateral intraocular disease with chemotherapy and focal therapy, a similar “conservative” approach may be appropriate in unilateral intraocular disease, especially in very young children who may later evidence contralateral disease.48 For patients with early intraocular disease (Reese-Ellsworth groups I to III, International Classification of Retinoblastoma group B), a nonintense two-drug regimen of vincristine and carboplatin had been suggested, based on overall results with three-drug chemotherapy (also including etoposide) documented at nearly 100% for ocular and visual preservation.28


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


