the absence of WTX, β-catenin fails to be phosphorylated, is stabilized, and accumulates in the nucleus, where it forms a complex with the Lef-1/TCF family of transcription factors to promote expression of growth-related genes.19
TABLE 29.1 Syndromes Associated with WT | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
in such families, the mode of inheritance is generally thought to be autosomal dominant, with variable penetrance and expressivity.24 Only one-tenth of the kindreds reported involve affected parents.23 More often, the disease occurs in siblings, cousins, or other relatives. A survey of 191 children of 99 patients with unilateral WT did not identify a single case of cancer.25
TABLE 29.2 Biologically Defined Subsets of WT | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
distinction of a pure rhabdomyoblastic WT (which is quite rare) from a primary renal rhabdomyosarcoma is often impossible on morphologic grounds. Finally, purely tubular and papillary WT may at times be difficult to distinguish from metanephric adenoma and papillary RCC.
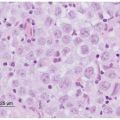
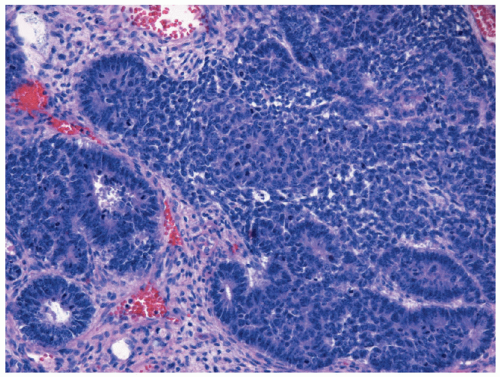 Figure 29.1 Triphasic WT, with well-defined tubules emerging from dense clusters of cohesive blastemal cells. Zones of pink-staining stromal differentiation separate blastemal nodules (H & E 100×). |
TABLE 29.3 The Revised SIOP Working Classification of Renal Tumors | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
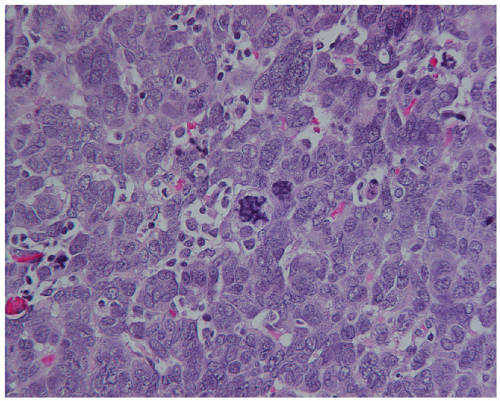 Figure 29.2 Anaplastic WT with a large, darkly stained multipolar mitotic figure, near the center, and a markedly enlarged interphase nucleus to its left. Nuclei throughout this field exhibit increased variation in size and shape (H & E 600×). |
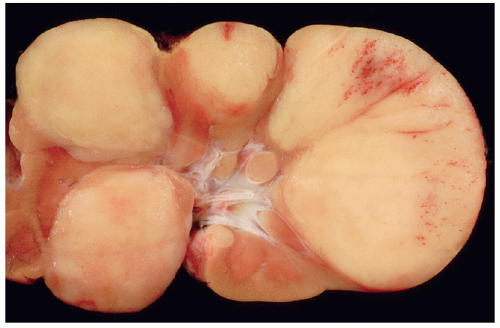 Figure 29.3 Perilobar nephrogenic rests. Grossly these PLNR are roughly wedge shaped following the contours of the renal lobule. The nephrogenic rest tissue is homogeneous and paler than the normal surrounding renal parenchyma. |
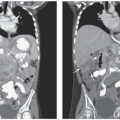
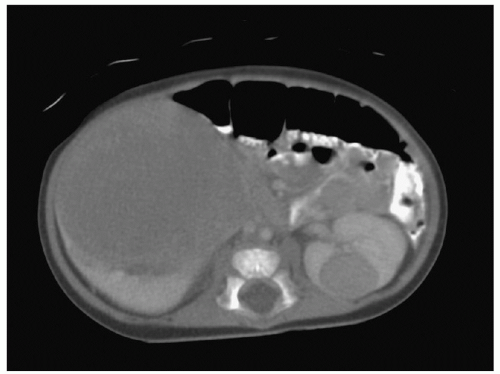 Figure 29.4 Axial CT scan of the abdomen demonstrating bilateral WTs. The larger right kidney tumor demonstrates the “claw” sign, in which the renal parenchyma is stretched around and cupping the tumor suggesting the organ of origin. |
classification is based on immediate nephrectomy and takes into account surgical-pathological findings with imaging for distant metastasis (Table 29.4). SIOP utilizes a preoperative chemotherapy approach, and staging criteria are based on a combination of prechemotherapeutic imaging to define metastatic disease and local operative findings following chemotherapy. Although the stage definitions are similar, the prechemotherapy stage does not have equivalent clinical significance to the postchemotherapy stage, thus confounding head-to-head comparisons of SIOP and NWTSG/COG trial results.
TABLE 29.4 Children’s Oncology Group Staging system for WT | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
|
Related posts:




Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


