Chapter 73 Rare Pediatric Tumors
Primary Liver Tumors of Childhood
Etiology and Epidemiology
Primary malignant liver tumors (PMLTs) of childhood are very rare malignancies, comprising between 0.5% and 2% of pediatric tumors.1 The incidence in the United States is approximately 1.6 per million.2 The most common forms are hepatoblastoma (HBL) and hepatocellular carcinoma (HCC), but benign vascular tumors, mesenchymal hamartomas, adenomas, nodular hyperplasia, and hepatobiliary tumors are reported as well.1 The median age at presentation for HBL is 1 year, and it predominates in males with a ratio of 1.5 : 1. HCC presents at a median age of 12 and is more prevalent in males with ratios varying from 2 : 1 to 11 : 1.
HBL has been associated with prematurity, low birth weight, Beckwith-Wiedemann syndrome, familial adenomatous polyposis, maternal ingestion of oral contraceptives, and fetal alcohol syndrome.3 HCC has a strong association with hepatitis B virus (HBV) and is also associated with α1-antitrypsin deficiency, hereditary tyrosinemia, extrahepatic biliary atresia, Fanconi anemia, ataxia-telangiectasia, Sotos syndrome, and glucose-6-phosphatase deficiency.
Biologic Characteristics and Molecular Biology
HBL can be associated with several genetic aberrations. These include Beckwith-Wiedemann syndrome, most prominently, with loss of heterozygosity (LOH) at 11p15, as well as familial adenomatous polyposis and Li-Fraumeni syndrome. The most common chromosome aberrations are extra copies of 1q, 2q, 7q, 8, 17q, and 20. LOH of 11p15 is seen in a third of patients with HBL, and LOH of chromosome 1p is seen in an additional third.3 Mutations of β-catenin and activation of Wnt/β-catenin signaling have been associated with HBL. Wilms’ tumor and rhabdomyosarcoma have also been associated with HBL.
Pathology and Patterns of Spread
HBL is the most common PMLT of childhood, representing 60% to 75% of cases.3 The most widely used classification system was proposed by Ishak and Glunz.3 It distinguishes two forms of HBL, the epithelial type and the mixed type. The epithelial type encompasses a poorly differentiated embryonal type and a highly differentiated fetal type. The pure fetal hepatoblastoma (PFH) has normal hepatocytes with rare mitoses and is associated with a favorable prognosis. Mixed-type HBL contains both epithelial elements and mesenchymal tissue. The small cell undifferentiated (or anaplastic) histologic subtype of epithelial HBL has a very poor prognosis.
HCC accounts for 25% to 40% of childhood PMLTs. There is a strong relationship with preexistent hepatic disease or cirrhosis. HCC is divided into traditional HCC and a fibrolamellar histologic variant. The fibrolamellar variant tends to occur in noncirrhotic livers of adolescents and is characterized by large polygonal neoplastic cells with lamellar collagen bundles. Historically, this variant was thought to have a higher resection rate and superior outcome compared with traditional HCC. However, a report from the Pediatric Intergroup Hepatoma Protocol INT-0098 found that fibrolamellar histology was not associated with superior response to standard therapies or prognosis.5
Clinical Manifestations, Patient Evaluation, and Staging
Workup begins with an ultrasound evaluation of the abdomen, which typically reveals a solid hepatic mass. Magnetic resonance imaging (MRI) and computed tomography (CT) are useful to delineate the mass, its vascularity, and the potential for resection. Surgeons may require an angiogram to evaluate the hepatic arteries before determination of resectability. Imaging of the chest with CT should be performed; lung metastases are apparent at diagnosis in 20% of children with HBL and 30% of children with HCC.6–8 Laboratory evaluation includes routine chemistries as well as determination of the serum alpha-fetoprotein level, which is elevated in over 90% of children with HBL and 60% to 80% of children with HCC.1,3,7–9 An alpha-fetoprotein value less than 100 ng/mL is associated with a poor prognosis.
The most common staging systems for PMLT are the PRETEXT presurgical system and the Children’s Oncology Group postoperative system (Table 73-1). The PRETEXT system (Fig. 73-1) was created in 1990 by the International Society of Pediatric Oncology Liver Study Group (SIOPEL) and is based on preoperative imaging and the number of affected liver segments.6
TABLE 73-1 Children’s Oncology Group Staging System for Hepatoblastoma
| Stage | Description |
|---|---|
| I | Completely resected localized tumors |
| II | Grossly resected tumors with microscopic residual tumor |
| III | Unresectable tumors (measurable residual tumor or abdominal lymph node involvement) |
| IV | Distant metastases |
From Schnater JM, Kohler SE, Lamers WH, et al: Where do we stand with hepatoblastoma? A review. Cancer 98:668-678, 2003.

Figure 73-1 PRETEXT staging system for hepatoblastoma.
Adapted from Schnater JM, Aronson DC, Plaschkes J, et al: Surgical view of the treatment of patients with hepatoblastoma. Results from the first prospective trial of the International Society of Pediatric Oncology Liver Tumor Study Group. Cancer 94:1111-1120, 2001.
Primary and Adjuvant Therapy and Results
The SIOPEL-1 trial reported by the International Society of Paediatric Oncology (SIOP) tested four to six courses of preoperative cisplatin and doxorubicin. Eighty-two percent of patients showed at least a partial response based on imaging and alpha-fetoprotein levels; 83% underwent delayed surgery, and in 77% of cases surgery achieved complete resection. The 5-year event-free survival and overall survival (OS), were 66% and 75%, respectively.8 The SIOPEL-3 trial compared cisplatin alone with cisplatin plus doxorubicin for three cycles preoperatively, followed by two postoperative cycles in children with HBL involving three or fewer liver segments with an alpha-fetoprotein level less than 100 ng/mL. Outcomes at 3 years were identical: 95% and 93% underwent complete resection in the respective chemotherapy arms, and OS was 95% and 93%, respectively. Grade 3 and 4 toxicities were significantly more common in the combination arm.10
Children with HCC on the SIOPEL-1 trial tended to have more advanced disease at presentation and fared significantly worse than patients with HBL. Partial response was seen in 49%, and complete resection was achieved in only 36%; 5-year OS and event-free survival were 28% and 17%, respectively.7 The Pediatric Intergroup Hepatoma Protocol INT-0098 randomized patients with HCC to postoperative cisplatin/vincristine/5-fluorouracil versus cisplatin and doxorubicin. There was no difference in results between the treatment regimens, and the overall 5-year event-free survival was 17% (75% for stage I, 8% for stage II, and 0% for stage IV).5 Subsequent SIOPEL studies evaluated a regimen of cisplatin and carboplatin, with no substantial improvement in outcome.7
Locally Advanced Disease and Palliation
Children with locally advanced and metastatic disease are treated with aggressive preoperative chemotherapy followed by surgery if possible. Pediatric Oncology Group study 9345 evaluated children with unresectable or metastatic HBL. Neoadjuvant therapy with carboplatin and carboplatin/vincristine/5-fluorouracil was followed by surgery when feasible or with high-dose cisplatin and etoposide. Resection was possible in 68% of patients with stage III disease and 36% of patients with stage IV disease; the 5-year event-free survival for the small number of children achieving resection was 79%.11 Current approaches include orthotopic liver transplantation when disease is unresectable, with documented long-term survival in a limited number of patients.6,11 The ongoing Children’s Oncology Group trial for locally advanced disease was highlighted previously; for “high-risk” patients with metastatic tumor or alpha-fetoprotein value less than 100 ng/mL, a national trial is testing the addition of irinotecan.
Metastatic disease is treated with systemic therapy followed by resection when possible. If local control can be achieved, metastasectomy is recommended. In rare patients with unresectable primary tumors, response to chemotherapy, and complete removal of pulmonary metastases, the SIOPEL-1 trial showed a limited number of children were long-term survivors.6
Irradiation Techniques
RT is not routinely used for PMLTs. An earlier series of 15 children with incompletely resected disease reported outcome after preoperative or postoperative irradiation.4 Doses ranged from 25 to 40 Gy in conjunction with chemotherapy. Six of eight patients treated postoperatively were disease free at 4 to 83 months, and one of four children with preoperative irradiation had histologic tumor control. The current Children’s Oncology Group HBL study specifically excludes use of RT. There are few current indications for hepatic irradiation or treatment beyond palliation to metastatic sites with HBL or HCC. RT has several technical challenges. Future attempts to incorporate RT for PMLTs in children would need to take advantage of modern radiotherapy techniques, including respiratory gating, given the critical organs surrounding the liver and the impact of combined therapeutic approaches.12
Pediatric Extracranial Germ Cell Tumors
Etiology and Epidemiology
Germ cell tumors (GCTs) are rare tumors in childhood, accounting for approximately 3% of pediatric malignancies.13–15 Teilium has described these tumors as arising from primordial germ cells that escape normal developmental influences. In the embryo, germ cells migrate from the allantoic stalk to the genital ridge and finally to the appropriate genital site. Therefore, GCTs can be found in the ovary or testis and aberrant migration accounts for the location of these tumors in midline structures such as the sacrococcygeal region, retroperitoneum, mediastinum, and pineal gland.
GCTs are more common in females than males.16 They have a bimodal age distribution, with extragonadal and testicular tumors occurring most frequently in children younger than 3 years old and gonadal tumors occurring most commonly in pubertal adolescents.
Prevention and Early Detection
GCTs are associated with intersex disorders, including pseudohermaphroditism, androgen insensitivity, and 5α-reductase deficiency. Undescended testis is highly associated with an increased risk of testicular malignancy, with the highest risk occurring in the intra-abdominal testis. This risk approaches 30 to 50 times the normal incidence of testicular cancer and can occur in either testis.14
Biologic Characteristics and Molecular Biology
Chromosomal abnormalities are common in malignant GCTs. The most frequent is isochromosome 12p [i(12p), which occurs in 75% to 80% of malignant ovarian or testicular GCTs]; other aberrations include loss of chromosome 13 and gain of chromosomes 21, 8, or 1q.17
Pathology and Pathways of Spread
GCTs span a spectrum of entities from benign to malignant, with elements of multiple histologic types in 25% of cases.16 The most common pediatric GCT is the teratoma, which is composed of tissue from more than one embryonic layer. Teratomas can be either mature, consisting of well-differentiated tissues, or immature, consisting of immature elements (most commonly neuroepithelial tissue).
Yolk sac or endodermal sinus tumors are highly malignant, occur most commonly in the ovary, testis, and sacrococcygeal region, and secrete alpha-fetoprotein. Histologically they show classic perivascular formations called Schiller-Duval bodies. Embryonal carcinomas are composed of large, pleomorphic undifferentiated cells. They often occur in combination with endodermal sinus tumors. Choriocarcinomas are rare tumors composed of malignant cytotrophoblasts and syncytiotrophoblasts. They typically secrete beta–human chorionic gonadotropin. All three of these GCT types are highly undifferentiated tumors with the propensity to metastasize to lung, liver, bone, and lymph nodes. In a series of 95 patients with sacrococcygeal endodermal sinus tumors,16 15% had positive lymph nodes at diagnosis and 35% had distant metastases.
Clinical Manifestations, Patient Evaluation, and Staging
The most common location for pediatric GCTs is the sacrococcygeal region, followed by the ovary, testis, and mediastinum. Unusual locations include the retroperitoneum, neck, stomach, and vagina. Sacrococcygeal tumors are classified according to Altman’s classification. Type I are predominately external, and type IV are entirely presacral. Neonates typically present with large, external, protruding sacral masses, which are commonly benign teratomas. Older children more commonly present with large pelvic masses, with malignant degeneration commonly apparent in children older than age 6 to 12 months.14
Initial evaluation should include a careful history and physical examination. Laboratory studies should include a complete blood cell count, renal and hepatic function tests, and evaluation of alpha-fetoprotein, beta–human chorionic gonadotropin, and lactate dehydrogenase levels; determination of CA-125 may also be of value, especially in sacrococcygeal teratomas. Tumor marker profiles for different histologies are listed in Table 73-2. Imaging includes ultrasonography of the primary site, CT of the abdomen and pelvis to evaluate for lymphadenopathy and resectability as appropriate, and CT of the chest to evaluate for metastatic disease. For secreting tumors, primary resection may be performed for both diagnosis and treatment if it is deemed that a complete resection can be performed. In nonsecreting and unresectable tumors, an open biopsy should be performed before starting definitive therapy.

GCTs are staged according to the site of origin using the Children’s Cancer Group and Pediatric Oncology Group staging system. The staging systems for ovarian, testicular, and extragonadal sites are outlined in Table 73-3.
TABLE 73-3 Children’s Cancer Group and Pediatric Oncology Group Staging System for Pediatric Germ Cell Tumors
| Stage | Description |
|---|---|
| Ovarian | |
| I | Limited to ovary (ovaries), peritoneal washings negative, tumor markers normal after appropriate half-life |
| II | Microscopic residual or positive lymph nodes (<2 cm), peritoneal washing negative, tumor markers positive or negative |
| III | Lymph node involvement >2 cm, gross residual disease, biopsy only, contiguous visceral involvement, peritoneal washings positive, tumor markers positive or negative |
| IV | Distant metastases |
| Testicular | |
| I | Limited to testis, tumor marker normal after appropriate half-life, completely resected with high inguinal orchiectomy |
| II | Transscrotal orchiectomy, microscopic disease in scrotum or high in spermatic cord, retroperitoneal lymph node <2 cm, increased tumor marker after appropriate half-life |
| III | Retroperitoneal lymph node >2 cm, no visceral of extra-abdominal involvement |
| IV | Distant metastases |
| Extragonadal | |
| I | Complete resection at any site, negative margins, coccygectomy for sacrococcygeal sites |
| II | Microscopic residual, lymph nodes negative |
| III | Gross residual or biopsy only, regional lymph nodes positive or negative |
| IV | Distant metastases |
Primary Therapy, Adjuvant Therapy, and Results
The optimal surgery for ovarian tumors has been debated, and historically principles of resection were based on the adult epithelial ovarian tumor experience. Recently the Children’s Oncology Group reported a series of children with ovarian GCTs who underwent conservative surgical resection and platinum-based chemotherapy; the 6-year survival rate approximated 95%. These researchers concluded that surgical guidelines should be changed to favor a more conservative approach including collection of ascites; examination and palpation of peritoneal surfaces, retroperitoneal lymph nodes, omentum, and the contralateral ovary with biopsy or excision of suspicious nodules; and complete resection of the ipsilateral tumor with potential sparing of a normal fallopian tube.18
Sacrococcygeal tumors are often resected using an inverted-V incision to spare the levator ani muscle and external sphincter. The entire coccyx must be resected because local recurrences of up to 37% are reported with inadequate surgery.15
Adjuvant therapy is based on the stage, degree of resection, and histologic subtype (Table 73-4). Currently there is no indication for adjuvant chemotherapy in mature or immature teratomas, and these patients are followed with close observation after surgery. Reported survival with surgery alone varies in the literature from 82% to 100%.14 In the Pediatric Oncology Group 9048/CCG 8891 trial, children with extracranial immature teratomas (32% with a malignant element) were treated with surgery alone with a 3-year overall event-free survival of 93%. Four of five patients with recurrence were disease free after platinum-based salvage chemotherapy.19
TABLE 73-4 General Treatment Guidelines for Extracranial Germ Cell Tumors
| Low-Risk Patients | |
| All teratomas | Surgery and observation |
| Stage I gonadal | |
| Stage I extragonadal | |
| Intermediate-Risk Patients | |
| Stage II-IV gonadal | Surgery with adjuvant chemotherapy |
| Stage II extragonadal | |
| High-Risk Patients | |
| Stage III-IV extragonadal | Surgery with adjuvant chemotherapy |
| Initial Unresectable or Biopsy Only | Neoadjuvant chemotherapy followed by second surgery, followed by adjuvant chemotherapy for residual pathologic disease |
There has been dramatic improvement in survival for children with stage II or greater malignant GCTs with the addition of cisplatin-based chemotherapy. The most commonly used regimen is PEB (cisplatin/etoposide/bleomycin). Using this regimen, OS has increased to more than 80% for all stages of GCTs.16,18,20,21
Locally Advanced Disease and Palliation
Bulky, locally invasive, and metastatic disease require treatment with neoadjuvant chemotherapy followed by second-look surgery; additional chemotherapy is utilized for residual tumor at the time of surgery. Even in this setting the OS is excellent owing to the chemosensitivity of these tumors. For high-risk patients, a high-dose PEB regimen (HDPEB) has been compared with standard-dose PEB. The Pediatric Oncology Group 9040/CCG 8882 trial20 randomized patients with stage III and IV gonadal and stage I to IV extragonadal GCTs to HDPEB versus PEB, reporting 6-year event-free survival of 89.6% versus 80.5% but no benefit in OS; there were more toxic deaths in the high-dose arm of the trial. Surgery for metastatic foci is usually not indicated because these foci tend to respond to chemotherapy. For relapsed or refractory tumors, chemotherapy with ifosfamide added to platinum and etoposide is recommended.
Irradiation Techniques
The role of RT for GCTs has diminished significantly owing to the effectiveness of current chemotherapy regimens. Historically, RT was used in conjunction with surgery and chemotherapy, with OS rates in the range of 48% to 62%.13,22,23 Seminomas and dysgerminomas were often cured with limited surgery and RT. Currently, the role of RT is limited to cases with unresectable, refractory, or recurrent disease unresponsive to chemotherapy. Classically, disease control for malignant GCTs has required radiation doses of 40 to 45 Gy or more for residual disease or disease that is resistant to chemotherapy.
Treatment Algorithms, Controversies, Challenges, and Future Possibilities
Treatment is based on tumor site, type, and grade and is detailed in Table 73-4. Surgical resection is necessary for local control and is preceded and/or followed by PEB chemotherapy. In patients with locally advanced or metastatic disease, survival still approaches 75% owing to the sensitivity of these tumors to platinum-based chemotherapy. With the high curability of these tumors, current protocols are using risk-adapted algorithms based on histology, site, stage, and genetic aberrations to decrease toxicity associated with chemotherapy.
Juvenile Nasopharyngeal Angiofibroma
Etiology and Epidemiology
Juvenile nasopharyngeal angiofibroma (JNA) is a highly vascular tumor that is histologically benign but locally invasive. JNA occurs almost exclusively in adolescent boys and young adult men, suggesting a prominent hormonal role in the tumor’s etiology; the average age at diagnosis is 17 years.24,25 There is an increased incidence of JNA in patients with familial adenomatous polyposis, suggesting a connection to the β-catenin pathway.26
Biologic Characteristics and Molecular Biology I
Both androgen and estrogen receptors have been demonstrated in JNAs.27,28 Recently, estrogen beta-adrenergic receptors have been found in a high percentage of JNAs. Schlauder and associates28 have postulated that the presence of aromatase in tumor cells converts endogenous androgens to estrogens, causing tumor growth via an autocrine-like mechanism.29 Most tumors stain for vascular endothelial growth factor.30
Pathology and Pathways of Spread
JNA is a benign tumor, although its exact nature is controversial. Some have suggested that it is a vascular hamartoma and similar to a hemangioma,31 but others believe it is neoplastic.32 Histologically, tumors are composed of fibrous connective tissue with abundant endothelium-lined vascular spaces.31 Localization of β-catenin to tumor stromal cells suggests these may be the neoplastic component rather than the endothelial cells.33 Tumors typically arise from the superior margin of the sphenopalatine foramen and invade laterally through the pterygomaxillary fissure toward the infratemporal fossa.24 Intracranial extension is seen in up to a third of cases, although actual dural invasion is uncommon.34,35 Tumors can be locally invasive of bone and extend into the parapharyngeal spaces, paranasal sinuses, orbit, and base of skull. This pattern of spread predicts for a high risk of local recurrence.35 Blood supply is primarily from the internal maxillary arteries of the external carotid system.
Clinical Manifestations, Patient Evaluation, and Staging
Presenting symptoms include recurrent painless spontaneous epistaxis, nasal obstruction, nasal discharge, a reduced sense of smell, snoring, headache, cranial nerve palsies, and facial swelling.25 Angiography is essential to define the tumor’s blood supply for planning surgery and, typically, for preoperative embolization to decrease surgical blood loss. There are usually multiple tortuous feeding vessels with a dense, homogeneous blush in the capillary phase.36 CT and MRI help define the anatomic extent of the enhancing tumor. Distant metastases do not occur, so systemic evaluation is not required. Biopsy can be hazardous owing to the tumor’s vascularity; not all authors require biopsy confirmation before treatment if clinical and radiographic data are consistent with the diagnosis.37
Several staging systems have been proposed (Table 73-5). Most are designed to guide decisions regarding the resectability and optimal surgical approach to the tumor rather than to predict prognosis.37,38–41
TABLE 73-5 Staging Systems for Juvenile Nasopharyngeal Angiofibroma
| Andrews Staging System39 | |
| Stage I | Tumor limited to the nasal cavity and nasopharynx |
| Stage II | Tumor extension into the pterygopalatine fossa, or maxillary, sphenoidal, or ethmoidal sinuses |
| Stage IIIa | Extension into the orbit or infratemporal fossa without intracranial extension |
| Stage IIIb | Stage IIIa with minimal extradural intracranial extension |
| Stage IVa | Extensive extradural intracranial or intradural extension |
| Stage IVb | Extension into cavernous sinus, pituitary, or optic chiasm |
| Carrillo Staging System37 | |
| Stage I | Tumor limited to nasopharynx, nasal fossae, maxillary antrum, anterior ethmoid cells and sphenoidal sinus |
| Stage IIa | Invasion to pterygomaxillary fossae or infratemporal fossae anterior to pterygoid plates, with major diameter <6 cm |
| Stage IIb | Invasion to pterygomaxillary fossae or infratemporal fossae anterior to pterygoid plates, with major diameter ≥6 cm |
| Stage III | Invasion to infratemporal fossae posterior to pterygoid plates or posterior ethmoid cells |
| Stage IV | Extensive skull base invasion >2 cm or intracranial invasion |
| Chandler Staging System40 | |
| Stage I | Tumor confined to the nasopharynx |
| Stage II | Tumor extending into the nasal cavity and/or sphenoidal sinus |
| Stage III | Tumor involvement of one or more of the maxillary or ethmoidal sinuses, pterygomaxillary and infratemporal fossae, and orbit and/or cheek |
| Stage IV | Tumor extending into the cranial cavity |
| Fisch Staging System38 | |
| Type I | Tumor limited to the nasopharynx and nasal cavity with no bone destruction |
| Type II | Tumors invading the pterygomaxillary fossa and the maxillary, ethmoidal, and sphenoidal sinuses with bone destruction |
| Type III | Tumors invading the infratemporal fossa, orbit, and parasellar region remaining lateral to the cavernous sinus |
| Type IV | Tumors with massive invasion of the cavernous sinus, optic chiasmal region, or pituitary fossa |
Primary and Adjuvant Therapy and Results
Surgical removal, often preceded by tumor embolization, is the primary treatment for JNA. A craniofacial approach is used for locally advanced tumors. Endoscopic techniques have less morbidity and are effective for earlier-stage lesions.24 Gross total removal is usually curative.34 A number of cases prove not to be amenable to complete resection; depending on case selection, there is a rate of local recurrence after surgery that approximates 20% to 40%, with most recurrences occurring in large, incompletely resected lesions.24,25 Moderate doses of RT may be indicated for postoperative residual tumor, although observation is more often considered because spontaneous involution of tumor is a well-recognized phenomenon.42,43 Most recurrences present within a year of surgery. The risk of recurrence is greatest in patients with large tumors that erode the skull base, young age at presentation, and irradiated tumors that are slow to regress.36,44 There are currently no indications for adjuvant chemotherapy after initial resection.
Locally Advanced Disease and Palliation
RT can provide effective control for recurrent or large, unresectable tumors. Objective tumor response after RT is typically slow, but ultimate control rates range from 75% to 92%45–48; 90% of responders have no residual tumor 3 years after treatment.49 Given the strong association of JNA with hormonal receptors, there has been interest in hormonal manipulation for patients with advanced disease. Diethylstilbestrol and flutamide have been used, but results have been inconsistent.36,50,51 Case reports of chemotherapy for recurrent tumor show efficacy for doxorubicin, dactinomycin, vincristine, cyclophosphamide, and cisplatin in selected patients.52,53,54
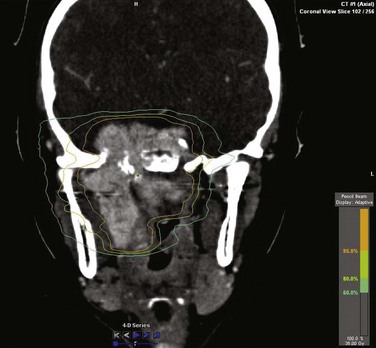
Techniques of Irradiation
No radiation dose-response curve has been reported for JNA. Local control rates greater than 80% are seen with doses ranging from 30 to 50 Gy,37,55,56–59 and a dose 36 Gy is commonly used. Intensity-modulated RT (IMRT) techniques provide high conformality for sparing of normal structures (Fig. 73-2). Stereotactic radiosurgery using single doses of 17 to 20 Gy and hypofractionated RT using 45 Gy in three fractions have been reported effective in small series of patients.60–62
Pleuropulmonary Blastoma
Etiology and Epidemiology
Pleuropulmonary blastoma (PPB) is a dysontogenetic neoplasm of childhood that originates in the lung and/or pleura.63 Although rare, it is the most common primary lung tumor in children.64 It is analogous to other dysontogenetic tumors such as Wilms’ tumor, neuroblastoma, and hepatoblastoma.65
PPB is thought to progress through a distinct sequence of clinical and pathologic changes beginning as a relatively nonaggressive cystic lesion and subsequently developing into the more malignant mixed cystic/solid and purely solid morphologies.66 Median age at presentation is 3 years.67 Younger children typically present with predominantly cystic tumors, whereas older children are more likely to have significant solid components.68 Boys and girls are equally affected.68 Siblings of patients with PPB have a higher incidence of PPB than the general population, although a genetic cause has not yet been found.69 There is also an increased incidence of other types of dysplasia and neoplasia in relatives of children with PPB.70,71
Prevention and Early Detection
Cystic PPB is clinically indistinguishable from benign lung cysts, and some authors recommend excision of all such lesions.69 Further supporting this approach is evidence that PPB can arise de novo from lung cysts, so that resection of these “precancerous” lesions is indicated.72,73 Others advocate a policy of watchful waiting for purely cystic lesions, only intervening if radiographic changes suggest progression.
Biologic Characteristics and Molecular Biology
TP53 mutations have been described in PPB and may portend a worse prognosis.74 Polysomy of chromosome 8 has also been noted.75
Pathology and Pathways of Spread
Histologically, PPB has small, primitive blastemal cells separated by an uncommitted sarcomatous stroma.65,68,76 Tumor cells stain with vimentin and may show myogenic differentiation.64 Rhabdomyoblasts and cartilage nodules are reported in 40% to 50% of patients.69
Tumors usually arise in the lung parenchyma. Spread to contiguous structures such as the mediastinum and pleura is associated with a poorer prognosis. Invasion into the chest wall is uncommon.77 Hematogenous metastases occur, most often to the brain. In one series,63 the incidence of brain metastases was 11% in mixed cystic/solid tumors and 54% in purely solid tumors.
Clinical Manifestations, Patient Evaluation, and Staging
Presenting symptoms of PPB include cough, dyspnea, wheezing, symptoms of respiratory infection, and, occasionally, spontaneous pneumothorax. Chest CT usually reveals a heterogeneous low-attenuation mass with pleural effusion and mediastinal shift.77 Differential diagnosis includes rhabdomyosarcoma, Askin tumor, and nonrhabdoid sarcomas; examination of the cystic fluid or solid tumor is required to make a diagnosis. Bone scintigraphy and MRI of the brain should be performed for staging, especially for tumors with a significant solid component.


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


