Radioimmunotherapy and Unsealed Radionuclide Therapy
 IMMUNOLOGY AND TARGETING CONSTRUCTS
IMMUNOLOGY AND TARGETING CONSTRUCTS
Immunity refers to protection from disease or infectious agents.1 Our immune system is composed of the cells and molecules responsible for the immune response, which can be divided into an early (1- to 12-hour) reaction, termed innate immunity, and a late (1- to >7-day) reaction, termed adaptive immunity. The innate immune system comprises biochemical and cellular mechanisms that exist prior to the introduction of a “foreign” or infectious agent and results in a rapid response. The innate immune system consists of epithelial barriers, phagocytic cells (neutrophils, macrophages), natural killer cells, the complement system, and cytokines. The adaptive immune system develops over time, becoming more effective with subsequent exposures of antigen. It exhibits the ability to “remember” and to respond more quickly with continued exposures to the same antigen. The adaptive immune system consists of lymphocytes and secreted antibodies. The adaptive immune system can be divided into humoral immunity and cell-mediated immunity. Concerning humoral immunity, B lymphocytes secrete antibodies for protection. Concerning cell-mediated immunity, helper T lymphocytes either activate macrophages or cytotoxic T lymphocytes, which then directly destroy pathologic (infectious or malignant) cells.
It is well known that the host’s immune system is important for preventing the growth and development of cancer.2 A large body of literature exists supporting the concept that the host immune system interacts with tumorigenesis and tumor progression. It has been shown in animal models and in the clinic that cancer immune surveillance is exceedingly important. For example, mice with an impaired innate or adaptive immune system will be more susceptible to develop chemically induced or spontaneous cancers. Additionally, the malignant transformation of cells in animals and humans, caused by the accumulation of somatic mutations and/or the deregulation of oncogenes or tumor suppressor genes, results in the expression of tumor antigens (TAs). These TAs are often recognized by the immune system as documented by TA-specific T-cell precursors and natural killer cells, found in the peripheral blood of cancer patients, capable of killing tumor cells. Further evidence of cancer immune surveillance exists in patients with genetic or drug-induced immunosuppression. Transplant patients exhibit a predisposition for certain malignancies (squamous cell carcinoma, basal cell carcinoma, Kaposi’s sarcoma, melanoma, and lymphoma). Patients with Chediak-Higashi and Wiskott-Aldrich syndrome demonstrate an increased rate of lymphoproliferative malignancies. Discontinuing immunosuppressive drugs in solid organ allograph patients with occult malignant melanoma has resulted in tumor regression.
Despite the evidence of cancer genesis and progression in immune-compromised hosts, the majority of cancers develop in seemingly immune-competent individuals. The last decade of research has revealed that cancer cells have developed means to avoid immune detection and surveillance, either through the selection of nonimmunogenic tumor cells and/or the active suppression of the immune response. It has therefore been rightfully suggested that “tumor immune escape” be added to Hanahan and Weinberg’s six hallmarks of cancer (self-sufficiency in growth signals, insensitivity to antigrowth signals, tissue invasion and metastasis, limitless replicative potential, sustained angiogenesis, and evasion of apoptosis).
TABLE 26.1 SELECT MONOCLONAL ANTIBODIES EVALUATED FOR RADIOIMMUNOTHERAPY
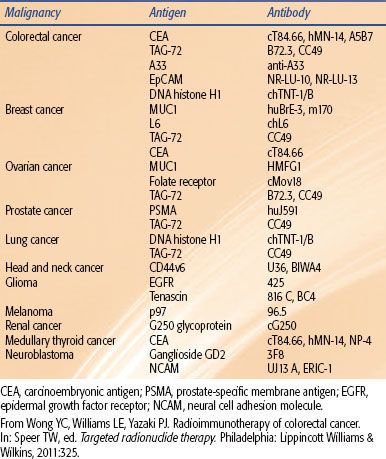
The targets for radioimmunotherapy (RIT) typically consist of tumor-associated antigens (TAAs). The reason for this is because the cytotoxic radionuclide must be delivered preferentially to malignant tissue and should avoid normal tissue. To date, >2,000 TAAs have been identified (http://www2.licr.org/CancerImmunomeDB). One of the main methodologies used to identify TAAs is termed SEREX (serologic analysis of recombinant cDNA libraries). SEREX involves a bacteriophage recombinant cDNA expression library, prepared from various malignancies (isolated tumors or malignant cell lines) or testis tissue.3 This cDNA expression library is transduced in Escherichia coli to produce a recombinant protein library. These various proteins (clones) are then tested against the serum from autologous cancer patients. Clones that react to IgG antibodies are identified and are then further characterized as TAAs. Many of these SEREX-identified TAAs have been elucidated by other processes and laboratories. This has led to the concept of a finite number of TAAs that are produced in cancer patients and are potentially identified by the immune system. These finite TAAs are collectively referred to as the cancer immunome. SEREX-defined antigens, representing broad categories, may be organized as follows: mutational antigens, amplified or overexpressed antigens, differentiation antigens, and cancer/testis antigens. Within these categories, only a limited number of TAAs have been used as targets for RIT (Table 26.1).
The ideal target for RIT targeting constructs would be one that is overexpressed on cancer cells, is uniformly expressed, is not found to any significant level in normal tissue, is not shed into the circulation, and exhibits an important role in tumor growth and progression.4 TAAs, as the name implies, are antigens “associated” with tumors but are also present in normal tissue. True tumor-specific antigens have not yet been identified and utilized. Overexpression is necessary because typical targeting constructs require antigen densities ≥105 receptors on each cell for adequate targeting. A homogenous antigen expression is desired so that a uniform activity distribution of the radionuclide will result. Nonuniform activity distributions (heterogeneity of antigen in target tissue being one potential cause) will significantly lower the effectiveness of RIT by subsequently resulting in nonuniform or heterogeneous dose distributions.5 This is particularly important for radionuclides with short path lengths of the emitted particles (i.e., Auger and α-particle emitters). Radionuclides with longer path lengths, such as high-energy β-emitters, can partly overcome the problem of nonuniform dose distributions through the crossfire effect. If the target antigen is significantly shed into the circulation, the targeting construct may bind and “complex” with the antigen. This will result in a more rapid clearance of the RIT agent and a much less effective treatment. If the TAA has an important signaling role, then subsequent binding of the targeting construct will most likely add to the cytotoxicity of the radionuclide because of the blockade or promotion of intracellular signaling, potentially resulting in disruption of growth pathways important for tumor growth. Some TAAs (receptors) will internalize when bound by the targeting construct. In truth, most receptors internalize, although they do so at different rates. A rapid internalization process will have an impact on the type of radionuclide that is selected and potentially on the delivery strategy of the RIT agent.
A multitude of agents have been used as carriers (targeting constructs) for the targeted delivery of radiation to cancer. These consist of antibodies, antibody fragments, peptides, affibodies, aptamers, and nanostructures (i.e., liposomes, nanoparticles, microparticles, nanoshells, and minicells). By an exceedingly large margin, intact monoclonal antibodies (mAbs) have dominated the field of RIT as targeting constructs6 (Fig. 26.1). In humans, there are five classes or isotypes of antibodies (IgA, IgD, IgE, IgG, and IgM). IgG is the most commonly used mAb for RIT because it is the most prevalent antibody in serum and has the longest serum half-life, typically measured in weeks (approximately 23 days). IgG is further divided into four subtypes, IgG1–4. IgG antibodies are large glycoprotein macromolecules, with an atomic mass of approximately 150,000 dalton (Da) or 150 kDa. The “y-shaped structure” (Fig. 26.1A) consists of two Fab fragments (fragment antigen binding; approximately 50,000 Da each) and an Fc fragment (crystallizable fragment; approximately 50,000 Da).
FIGURE 26.1. Antibody engineering: strategies to improve the therapeutic index in radioimmunotherapy. A: Typical structure of a humanized IgG antibody. The following engineering strategies are presented: Red dots, mutations in amino acids involved in FcRn binding that influence the pharmacokinetics of the IgG; CDR1-3, murine CDRs grafted into a human IgG backbone to humanize the antibody; Cys, engineered cysteine residues for site-specific conjugation. B: Humanization strategies: purple, indicating the murine portion of the IgG, and blue, indicating the human portion of the IgG (C) introducing a nuclear localizing signal (D) monospecific and bispecific fragments used in radioimmunotherapeutic strategies. CDR, complementarity-determining regions; CH, constant domain heavy chain; CL, constant domain light chain; Fc, crystallizable fragment; Fv, variable fragment. (From Burvenich IJG, Scott AM. The delivery construct: maximizing the therapeutic ratio of targeted radionuclide therapy. In: Speer TW, ed. Targeted radionuclide therapy. Philadelphia: Lippincott Williams & Wilkins, 2011:238.)
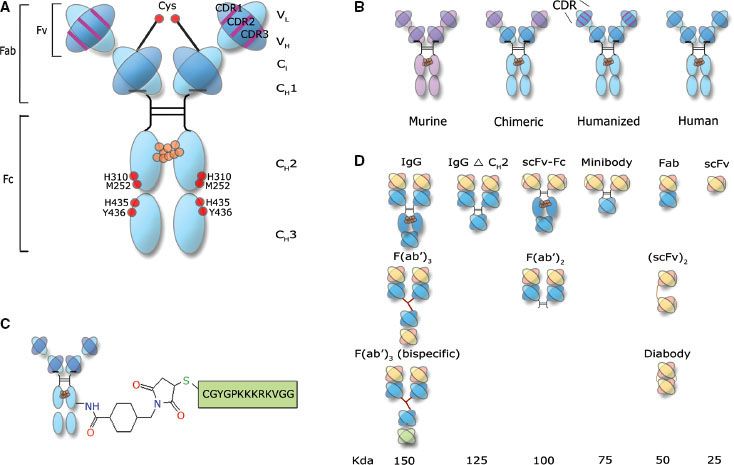
The “tip” of each Fab fragment has a variable amino acid sequence, from one mAb to another. Accordingly, each tip is an antigen binding site (ABS) and is responsible for antigen recognition. Each ABS forms a noncovalent bond (electrostatic forces, van der Waals forces, hydrophobic interactions, and hydrogen bonds) with the target or antigen. The specific region of an antigen, which binds to the ABS, is referred to as an epitope. It has been proposed that a million or more different antibodies exist in various individuals. Theoretically, >109 different antibodies can be produced. The outer core of the mAb consists of two identical light chains (outer portion of the Fab fragment) designated with an “L.” The inner core, consisting of the Fc region and the inner Fab region, is designated as heavy or “H.” Both the light and heavy chains contain homologous, 110 amino acid sequences that fold on one another and are connected by a disulfide bridge, resulting in “globular” motif or loop, called an Ig domain. There are three constant heavy domains (CH1-3) and only one constant light (CL), one variable heavy (VH), and one variable light (VL) domain. The ABS consists of a VL and a VH region. Within each variable domain, there are three hypervariable regions (about 10 amino acid residues per hypervariable region) that form a three-dimensional surface that is “complementary” to the shape of the antigen surface; they are called complementarity-determining regions (CDRs). A total of six CDRs come together to form the ABS. There are two ABS for each IgG mAb; hence, each IgG mAb is considered bivalent.
Affinity refers to the strength of the bond between the ABS and the antigen. The strength of this bond is represented by the dissociation constant (Kd). Avidity refers to the overall strength of the ABS-antigen interaction, depending on both the affinity and the valency of the interaction. It should be noted that a high-affinity interaction can improve specific delivery of the RIT agent and reduce overall dosing requirements. Increasing the affinity indefinitely, however, may decrease tumor penetration. It has been demonstrated that an affinity of 10–7 to 10–8 M is needed for tumor retention, whereas affinities in the range of ≥10–10 to 10–11 M will result in retention in normal tissue and asymmetric binding in tumor tissue, termed the binding site barrier.7 The binding site barrier phenomenon may be at least partially overcome by increasing the antibody mass, or the overall delivered quantity of antibody.
Unconjugated antibodies—those not attached to a radionuclide or cytotoxic agent—will also mediate biologic activities. These activities may be mediated by the Fc region of the mAb or may be Fc independent. Fc-mediated interactions are termed effector functions and consist of antibody-dependent cell-mediated cytotoxicity (ADCC) and complement-dependent cytotoxicity (CDC).7 Concerning ADCC, interaction of the Fc region of the antibody with Fc receptors (located on immune effector cells) results in the subsequent phagocytosis or lysis of the antibody-bound cancer cell. CDC is initiated by the interaction of soluble blood proteins and the Fc region. Epitope-dependent (Fc-independent) functions of the mAb may result in the inhibition of ligand binding, inhibition of ligand-induced dimerization, and inhibition of receptor shedding. These epitope-dependent functions are characteristic of modern-day biologics that target growth factor receptors, such as cetuximab and trastuzumab.
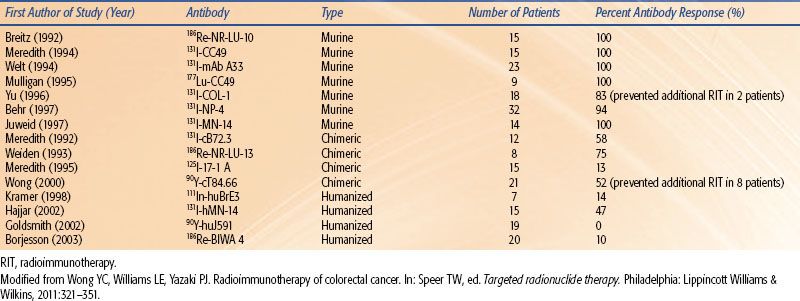
The original technology used to produce mAbs was first published by Kohler and Milstein8 in 1975 and is referred to as the hybridoma technique. The technique has propagated the use of murine mAbs for research and for therapy in the clinic. In fact, the two U.S. Food and Drug Administration (FDA) RIT agents used to treat non-Hodgkin lymphoma (NHL; ibritumomab tiuxetan and tositumomab) are murine mAbs. Although these agents are delivered as single instillations in patients typically with decreased immune recognition capabilities, there is a concern that human antiglobulin antibodies (HAGAs) will develop. If this phenomenon occurs in response to murine antibodies, then the resulting HAGAs will be called human antimouse antibodies (HAMAs). The formation of HAMAs will expedite blood clearance of the antibody and decrease targeting capabilities as well as potentially cause various adverse symptoms. Two main strategies, through the use of genetic engineering, have emerged6 that reduce the immunogenicity of mAbs: (a) the production of antibody chimeras derived from both murine and human DNA and (b) the production of humanized or fully human antibodies (Fig. 26.1B). Chimeric antibodies retain murine VH and VL domains, whereas humanized antibodies retain murine CDRs. Fully human antibodies retain no murine components. Although the development of HAGA may not be important after a single dose of mAb in lymphoma patients, its induction will have a greater detrimental impact for patients with solid tumors when treated with RIT.4 As can be seen in Table 26.2, as the targeting construct moves from a murine to humanized forms, the immunogenicity is lessened. This concept is important to employ multiple doses or fractions of RIT agents. Figure 26.1C illustrates the concept of adding a nuclear localizing signal to bring the mAb from the cell surface or cytoplasm into the cell nucleus.
TABLE 26.2 ANTIBODY IMMUNOGENICITY FOLLOWING A SINGLE ADMINISTRATION (SELECT SOLID TUMOR RADIOIMMUNOTHERAPY TRIALS)
Another factor that is critical and influences antibody targeting and pharmacokinetics is antibody molecular size (Fig. 26.1D). As stated previously, RIT has been less successful for treating solid tumors than hematologic malignancies. This is largely because of the lack of radiosensitivity of epithelial tumors (compared to hematologic malignancies) and the poor penetration of mAbs into large tumors. The decreased penetration of 150 kDa antibodies into large tumors is a direct result of increased tumor interstitial pressure, an aberrant tumor vasculature, and an abnormal tumor extracellular matrix.9,10–12 Additionally, 150 kDa antibodies need longer periods of time to accrete into tumors and have long serum half-lives. When radiolabeled, a long serum half-life of the targeting construct will increase exposure of the bone marrow to radiation, which causes hematologic toxicity and limits the amount of antibody and radionuclide that can be given. To overcome some of these issues, methods have been used to generate antibody fragments of varying size and valency. These smaller fragments exhibit superior tumor penetration and clear more rapidly from the circulation. However, if clearance from the circulation is too rapid, this can further limit tumor penetration. Table 26.3 summarizes these general concepts for targeting constructs of various molecular weights.
Although mAbs and their fragments represent the most commonly used targeting constructs for the delivery of a radionuclide to malignant tissue, other agents are either in use or are being investigated, consisting of peptides,13,14,15,16 affibody molecules,17,18–19 and aptamers.20,21–23 Nanostructures are also being investigated as carriers of radionuclides.24,25–26,27 In their unmodified form, the targeting capabilities of nanostructures are rather nonspecific.24,28
Peptides are small amino acid sequences (typically 7 to 14 amino acids) that serve as opioids, hormones, sweeteners, protein substrate inhibitors, releasing factors, antibiotics, and cytoprotectors.29 The overexpression of receptors that are specific for various peptides has led to the development of peptide-based radiopharmaceuticals.30 Somatostatin is one of the most common peptides and is overexpressed in a multitude of malignancies, including breast cancer, small cell lung cancer, medullary thyroid cancer, and neuroendocrine tumors (NETs). Somatostatin is rapidly degraded; however, its derivative, octreotide, is very stable. Octreoscan (indium-111 diethylenetriamine pentaacetic acid [111In-DTPA]) has been shown to be highly diagnostic for NETs. Affibody molecules17 are classified as affinity ligands or scaffold proteins that are approximately 7 to 9 kDa. These proteins are based on a 58 amino acid residue derived from staphylococcus protein A, which binds immunoglobulin. Various applications have been applied to affibody use, including radiolabeled targeting for therapy. Aptamers are small (8 to 12 kDa; 10 to 100 bases) single- or double-stranded oligonucleotides that are selected in vitro from a random library termed SELEX (systemic evolution of ligands by exponential enrichment). Aptamers are an attractive alternative to larger mAbs because they are easy to produce, have a low cost of production, exhibit high affinities, have a small size, are rapidly cleared from the circulation, have an unlimited shelf life, and are of low immunogenicity. Their major detriment is a short serum half-life (measured in minutes to hours) in their unmodified form. Aptamers are amazingly versatile and can recognize nearly any type of target, from metal ions to whole cells and even entire organisms. A complete database of known aptamers can be found at the Ellington Lab Web site (http://aptamer.icmb.utexas.edu). The current lack of radiolabeled aptamers is simply a portrayal of a very promising technology in its infancy.
 THE PHYSICS AND RADIOBIOLOGY OF RADIOIMMUNOTHERAPY
THE PHYSICS AND RADIOBIOLOGY OF RADIOIMMUNOTHERAPY
RIT delivers radiation to the target tissue in a continuous, although declining, low dose rate (LDR) fashion. Typical dose rates for RIT are in the range of 10 to 20 cGy per hour. The total dose delivered by RIT is low, in the range of 1,500 to 2,000 cGy,31 with an effective half-life of 24 to 72 hours. This can be compared to the high dose rate (HDR) delivery of radiation by external-beam radiation therapy (EBRT). EBRT typically will deliver radiation at a dose rate of 100 to 500 cGy per minute. It should be noted that these total dose ranges for RIT occur despite overall very low percent injected doses (0.1% to 10.0%) that ultimately localize in target tissue.32 Regardless, radiation-induced apoptosis is still induced.
TABLE 26.3 TARGETING AND PHARMACOKINETICS OF INTACT IGG AND VARIOUS ANTIBODY FRAGMENTS

FIGURE 26.2. Cell survival curves following treatment with radiotherapy. The blue curve represents low dose rate radiotherapy; the green curve represents high dose rate radiotherapy. (From Bernhardt P, Speer TW. Modeling the systemic cure with targeted radionuclide therapy. In: Speer TW, ed. Targeted radionuclide therapy. Philadelphia: Lippincott Williams & Wilkins, 2011:265.)
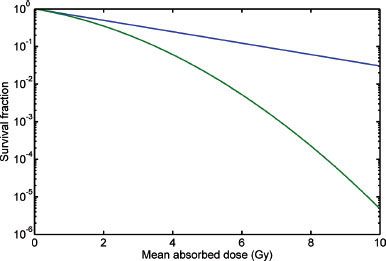
The most radiosensitive component of a cell is the DNA.33 Irradiation of tissue results in DNA damage. This damage may be either repaired or result in permanent damage. Permanent damage will cause cell death. By using a target-hit model, the tissue response end point of cell death may be used to relate absorbed dose of ionizing radiation to cell death. When the log surviving fraction of irradiated cells is plotted on the ordinate and the dose (Gy) is plotted on the abscissa, a cell survival curve is generated (Fig. 26.2). The “hit” that results in most lethal events is a double-strand break (DSB) of DNA. The mathematical term, α, represents the initial slope of the cell survival curve. It is a constant for a given tumor (or tissue) and can be thought of as the probability, per unit of absorbed dose, of creating a lethal DSB.34 The target is the resulting DSB, and the cell survival versus absorbed dose is a pure exponential function:

where S is the surviving cell fraction and D is the mean absorbed dose. Ionizing irradiation may also cause nonlethal single-strand breaks (SSBs). If these events accumulate, they may become lethal. The constant, β, is used to describe this phenomenon and represents the more distant, “linear” portion of the cell survival curve. The linear-quadratic (LQ) model combines the two processes into a continuously bending curve:

The shoulder on the cell survival curve is typically observed when HDRs of radiation are employed (green line in Fig. 26.2). In RIT, the dose rate is 1,000-fold lower; therefore, the quadratic portion of the curve will have a much lower impact on survival because many SSBs, considered sublethal damage, will be repaired during the more lengthy delivery of LDR radiation. This will result in a “small” or absent observable shoulder. Thus, when estimating cell survival for RIT, α alone will define the radiosensitivity of the tumor (blue line in Fig. 26.2). Considering dose rate, RIT is approximately 20% less effective than HDR EBRT.31 RIT, however, does appear to be relatively effective. This phenomenon can be attributed to many radiobiologic processes that appear to cause greater than predicted rates of apoptosis. These processes include low-dose/dose rate apoptosis, low-dose hyperradiosensitivity-increased radioresistance, inverse dose rate effect (G2 synchronization), radiation-induced biologic bystander effect, and the crossfire effect.32
FIGURE 26.3. Tumor control probability (TCP) for various radionuclides. TCP = 0.9 versus tumor mass. The optimal TCP for various tumor masses when treated with 211At, 177Lu, 131I, and 90Y. This corresponds to approximately 10–5, 10–2, 0.1, and 10 g, respectively. (From Bernhardt P, Speer TW. Modeling the systemic cure with targeted radionuclide therapy. In: Speer TW, ed. Targeted radionuclide therapy. Philadelphia: Lippincott Williams & Wilkins, 2011:266.)
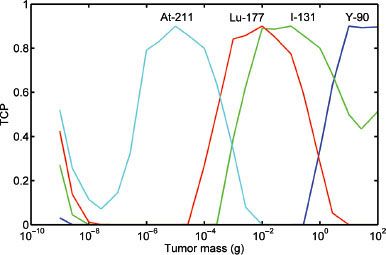
TABLE 26.4 COMMONLY USED RADIONUCLIDES FOR RADIOIMMUNOTHERAPY
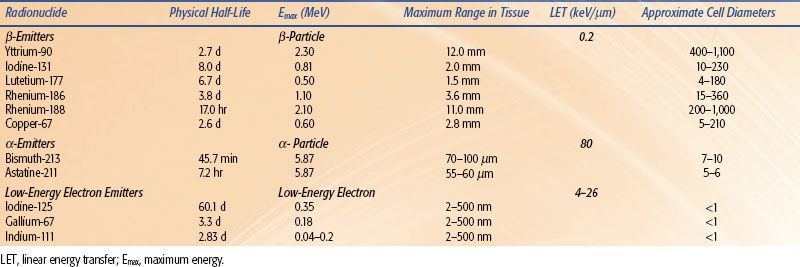
Various radionuclides have been used for RIT (Table 26.4), and their physical properties have been extensively reviewed in the nuclear medicine literature. They can be grouped into three basic categories depending on the type of emitted particulate radiation. Radionuclides that emit high-energy electrons are referred to as β-emitters. These electrons have maximum path lengths in tissue from 1.5 to 12.0 mm. This translates into a range of approximately 130 to 1,100 cell diameters. The most commonly used β-emitters for RIT are yttrium-90 [90Y] and iodine-131 [131I]. The maximum range of electrons in tissue for 90Y and 131I is 12 mm and 2 mm, respectively. It should be noted however, that 90% of the electron energy is deposited over 5.2 mm for 90Y and 0.7 mm for 131I. This range of 90% energy deposition is referred to as the R90. The most commonly used α-emitter for RIT is 211At. An α-particle is a helium nucleus that has a maximum range in tissue of 55 to 100 μm (5 to 10 cell diameters). Although it has a short range, the α-particle is very destructive and has a high linear energy transfer (LET). Low-energy electron emitters also emit radiation that is high LET and have path lengths between 2 and 500 nm (width of a double strand helix). Auger emitters, such as 111In or iodine-125 [125I], are most effective if delivered to the nucleus of a cell or incorporated into the DNA.
Because radionuclides have different energy spectra for their emitted particulate radiation, they will each interact with tissue and deposit their energy over varying distances. There is therefore a relation between type of radionuclide, tumor size, absorbed dose, and ultimately tumor cure probability (TCP). If it is assumed that a tumor has a spherical volume and contains a uniform and identical activity concentration of a radionuclide, then the TCP can be calculated for different radionuclides and tumor size.35 Figure 26.3 illustrates the relation between tumor mass and TCP for astatine-211 [211At], lutetium-177 [177Lu], 131I, and 90Y. As can be seen, there is an optimum tumor size for the different energy spectra for each radionuclide such that the TCP is maximized. If the tumor is small relative to the emission range, then much of the energy will be lost to the surrounding tissue and the absorbed dose will be low. As the tumor size increases, more energy is absorbed until the maximum TCP is reached. As the tumor further increases in size, the absorbed energy remains high, although fewer cells are affected by the radiation and TCP begins to decrease.35
Labeling the targeting construct with the appropriate radionuclide (radiochemistry) is exceedingly important and equally complex. Radionuclides are attached to targeting constructs by either using a “linker” molecule, termed a bifunctional chelating agent (BCA) or by a chemical reaction that forms a covalent bond between the radionuclide and the targeting construct. Three basic scientific fields converged to make radiochemistry a reality: coordination chemistry, directed biologic targeting, and the medical application of radiopharmaceuticals.36 In general, metallic radionuclides will require a BCA for labeling, and radiohalogens will require a chemical reaction (halogenation). The most prevalent therapeutic radionuclides used in RIT are 90Y (metallic radionuclide) and 131I (radiohalogen). One of the most commonly used BCAs is DTPA—a polyaminopolycarboxylate straight chain ligand. Tiuxetan, a modified DTPA molecule, is used as a linker molecule to chelate 90Y to ibritumomab (90Y ibritumomab tiuxetan; Zevalin, Spectrum Pharmaceuticals, Inc., Irvine, CA). Tiuxetan forms a urea-type bond37 to the antibody (ibritumomab), and its five carboxyl groups interact with and chelate 90Y to form a stable coordination sphere. The halogenation reaction that bonds 131I to the targeting construct (131I tositumomab; Bexxar, GlaxoSmithKline, Philadelphia, PA) is called iodination. Although there are many permutations of the iodination reaction, it basically inserts 131I into a tyrosine group on the mAb without the need for a chelation molecule. Regardless of the required labeling technique, it is incumbent that a reasonably high labeling yield, unaltered biodistribution, stability of the radionuclide, and immunoreactivity are preserved.
To date, a single instillation or fraction of the RIT agent is delivered systemically (i.e., Zevalin and Bexxar). It is well known that although relatively effective for hematologic malignancies, RIT is much less effective for treating solid tumors. Therefore, a number of strategies are being developed that will potentially increase the effectiveness of RIT. These strategies include modulating the tumor microenvironment, using pretargeting techniques, extracorporeal delivery, combined modality therapy (CMT), fractionation, multiple radionuclides (radionuclide cocktail), increasing antibody mass (the amount of antibody delivered systemically), alteration of the physical properties (size and affinity) of the targeting construct, and employing different types of LET radiation (i.e., b-emitter vs. α-emitter). All in all, these strategies are designed to—either alone or in combination—deliver more radiation to the tumor, make the radiation more cytotoxic, or decrease the exposure of radiation to bone marrow. As a result, the tumor to blood ratio will increase, and ultimately the therapeutic ratio will increase. The pretargeting strategy warrants further discussion.38–40,41
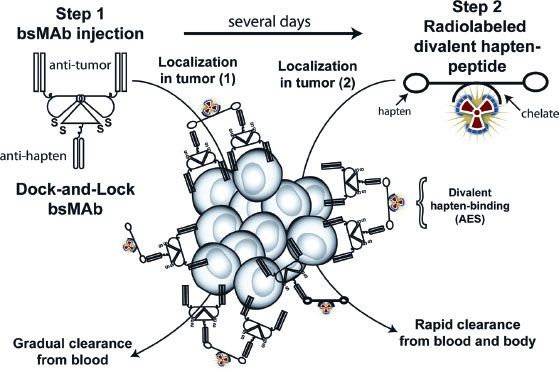
Because radiolabeled mAbs take 2 to 3 days to localize or accrete into tumors, antibody-based RIT results in a prolonged exposure of the bone marrow to radiation, causing hematologic toxicity and rendering the bone marrow as the dose-limiting normal tissue. Accordingly, the tumor/blood ratios of mAb will only slightly favor the tumor. This situation can seriously limit the successful prospects of antibody-based RIT, especially for treating solid tumors. Truly, smaller targeting constructs (antibody fragments) can be used for RIT, and they will exhibit pharmacokinetics that result in a more rapid blood clearance, allowing for the administration of higher activities. Unfortunately, because of the lower overall tumor accretion and retention of antibody fragments, the advantage of a more rapid blood clearance is usually offset. Therefore, the ideal delivery construct would manifest the targeting properties of an intact mAb but exhibit the blood clearance pattern of a small molecular weight construct. Because no known such construct exists, pretargeting strategies have been developed. The basic premise of pretargeting is to separate the delivery of a large, macromolecule-targeting construct (prolonged circulation time) from the delivery of a much smaller cytotoxic radioconjugate (more rapid circulation time). Two main approaches have been employed: a bispecific monoclonal antibody (bsmAb) system and a streptavidin-biotin system. In the bsmAb system (Fig. 26.4), a portion of the antibody has affinity for the tumor (antitumor), and another portion has affinity for the radionuclide carrier ligand or hapten-peptide (antihapten). Initially (step 1), a large “saturation” dose of the unlabeled bsmAb is administered, and the antibody localizes in the tumor over several days. Occasionally, a clearing step is used to facilitate the clearance of the bsmAb from the circulation. Subsequently (step 2), a radionuclide conjugated to a hapten-peptide is administered that has high affinity for the antihapten portion of the bsmAb. This step results in a rapid distribution of the radionuclide in the tumor owing to the high affinity of the hapten-peptide for the bsmAb. Because the hapten-peptide has a small molecular weight, it will clear rapidly from the body and result in a low bone marrow exposure to radiation.42 In the streptavidin-biotin system, streptavidin is conjugated to the initial pretargeting macromolecule, and biotin is conjugated to the radionuclide. Streptavidin and biotin have a very high affinity for each other (1015 M–1). When either system is used, the tumor/blood ratios of the targeting agent are significantly increased.43,44
FIGURE 26.4. Bispecific pretargeting procedure. The bsMAb is injected, and over several days it will localize in the tumor and clear from the blood. The bsMAb shown in this example is based on the dock-and-lock method for preparing recombinant bsMAb that has two binding arms for the tumor and one for the hapten. Once the molar concentration of the bsMAb is low enough, the radiolabeled hapten-peptide is given. The hapten-peptide has two haptens for more stable binding within the tumor, perhaps by cross-linking two adjacent bsMAb through a process known as the affinity-enhancement system (AES). The peptide portion usually contains four to five D-amino acids with a single chelator bound to one of the amino acids that is used to capture the radionuclide. (From Sharkey RM, Goldenberg DM. Pretargeted radioimmunotherapy. In: Speer TW, ed. Targeted radionuclide therapy. Philadelphia: Lippincott Williams & Wilkins, 2011:194.)
 CONJUGATED THERAPY
CONJUGATED THERAPY
To date, considerable progress has been made in the field of RIT for many different malignancies including NHL, Hodgkin lymphoma, leukemia, multiple myeloma, colorectal carcinoma, hepatocellular carcinoma (HCC), central nervous system (CNS) malignancies, medullary thyroid cancer, ovarian cancer, head and neck cancer, prostate cancer, renal cell carcinoma, osteosarcoma, NETs, melanoma, and pediatric malignancies. Although promising data is emerging for most disease sites, the hematologic malignancies appear to be the most radioresponsive. Progress has been less sanguine for solid tumor malignancies, and phase III trials are lacking.45,46–47,48–49 For the sake of clarity and brevity, this section will focus only on clinically relevant phase II/III trials and U.S. FDA (or its international equivalent)-approved RIT therapeutics.
Hematologic Trials and Approved Therapeutic Agents
Currently, there are two U.S. FDA-approved RIT agents in the United States: 90Y ibritumomab tiuxetan (Zevalin; 2002) and 131I tositumomab (Bexxar; 2003). Zevalin has U.S. FDA approval for relapsed or refractory follicular NHL and as a frontline adjuvant agent for follicular NHL achieving a complete response (CR) or partial response (PR) to induction chemotherapy. Bexxar has U.S. FDA approval for the relapse or refractory setting as well as transformed NHL. Both are murine IgG mAbs that target the CD20 surface antigen on follicular NHL.50 90Y ibritumomab tiuxetan utilizes 90Y, a pure b-particle emitter with a physical half-life of 2.7 days. The b-particle has an energy of 2.3 MeV and a maximum tissue penetration of approximately 12.0 mm (R90 = 5.2 mm). Tiuxetan is a DTPA-type chelate that attaches 90Y to the mAb, ibritumomab. Because there is no gamma emission in the spectrum of this isotope, it is not visualized by gamma camera scans. As a result, a biodistribution assessment cannot be performed. Therefore, a surrogate imaging radionuclide that emits gamma radiation (111In) is required. In contrast, 131I tositumomab is a mixed b/gamma emitter. The gamma spikes at 364 keV, and the beta emission has energy of 0.6 MeV. The maximum range in tissue of the b-particle is 2.3 mm (R90 = 0.7 mm). This agent can be imaged on gamma camera to calculate total body clearance. Although theoretical arguments can be rendered as to why one or the other therapeutic agent may have an advantage based on physical characteristics of the emission spectra, there is no convincing evidence that either Zevalin or Bexxar provides a clinical benefit for treating follicular NHL over each other, and they both appear to be mutually supportive.
For both agents, the treatment is delivered over 1 to 2 weeks. On day 1, both protocols deliver an infusion of nonradioactive (cold) anti-CD20 antibody (Zevalin employs rituximab; Bexxar employs tositumomab) designed to saturate the CD20 antigen sink (depletion of peripheral B-cells and the binding of nonspecific sites in the liver and spleen) and provide antibody mass, which improves biodistribution and tumor targeting.51,52 The administered activity for Zevalin is based on weight (0.4 mCi/kg for a platelet count ≥150,000; 0.3 mCi/kg for a platelet count of 100,000 to 149,000; maximum of 32 mCi). A single gamma scan (111In ibritumomab tiuxetan) is used to confirm a normal biodistribution on days 3 to 4. A review of the Zevalin imaging registry reveals that only 0.6% of scans exhibited an altered biodistribution. The administered activity for Bexxar is based on a calculated total body clearance (three scans over 1 week) that delivers a total body (red bone marrow) dose of 75 cGy. This calculation is reduced to a total body dose of 65 cGy for a platelet count <150,000. Eligible patients for both Zevalin and Bexxar are also required to have an absolute neutrophil count (ANC) ≥1,500 and a bone marrow biopsy that reveals <25% involvement with lymphoma.
Relapse Setting
Table 26.5 summarizes significant prospective phase II/III clinical trials providing evidence for the use of RIT for treating relapsed or refractory follicular NHL. Together they represent >200 patients treated with either Zevalin or Bexxar. Both agents appear to suggest an overall response rate (ORR) of 60% to 80% and a CR rate of 20% to 50%.
A phase III study comparing Zevalin versus rituximab for patients with relapsed or refractory low-grade follicular B-cell NHL or transformed NHL was performed.53 Patients were randomized to either a single intravenous (IV) dose of Zevalin 0.4 mCi/kg (n = 73) or IV rituximab 375 mg/m2 weekly for four doses (n = 70). The RIT group was pretreated with two rituximab doses (250 mg/m2) to improve biodistribution and tumor targeting. After the first rituximab dose on day 1, 111In ibritumomab tiuxetan was administered to assess biodistribution and to aide in dosimetry. No patients received the therapeutic dose of 90Y ibritumomab tiuxetan (Zevalin) if >20 Gy or 3 Gy was calculated to any nontumor organ or the red marrow, respectively. Zevalin was administered after the second rituximab dose approximately 1 week (days 7 to 9) after the first dose of rituximab and 111In ibritumomab tiuxetan. The administered activity of Zevalin was capped at 32 mCi. Patients in both arms of the study received two prior chemotherapy regimens. The ORR was 80% for Zevalin and 56% for rituximab (p = 0.002). The CR rates were 30% and 16% (p = 0.04), respectively, in the Zevalin and rituximab group. Durable responses ≥6 months were 64% versus 47% (p = 0.030) for Zevalin versus rituximab. The conclusion of the study was that RIT with Zevalin was well tolerated and resulted in statistically significant and clinically significant higher ORRs and CRs than rituximab alone.
TABLE 26.5 SUMMARY OF MAJOR PUBLISHED TRIALS OF RADIOIMMUNOTHERAPY FOR RELAPSED OR REFRACTORY LOW-GRADE NON-HODGKIN LYMPHOMA

In a pivotal, nonrandomized, phase III multicenter trial, patients with relapsed, refractory, or transformed follicular B-cell NHL were treated with Bexxar (n = 60), and the outcome was compared to patients’ last qualifying chemotherapy (LQC) regimen (n = 28).54 Eligible patients were required to have been treated with at least two prior protocol0specific chemotherapy regimens (median of four regimens in the study) and to either have not responded or progressed within 6 months of therapy. A PR or CR was observed in 39 patients (65%) after Bexxar compared to 17 patients (28%) after LQC (p <0.001). The median duration of response was 6.5 months for Bexxar and 3.5 months for the LQC group (p <0.001). The CR rate was 20% for Bexxar and 3% for the LQC group (p <0.001). The conclusion of the study was that a single dose of Bexxar was significantly more efficacious than the LQC received by heavily pretreated patients with relapse or refractory follicular B-cell NHL.
Frontline Therapy
Considering the concerns about RIT for treating large bulky tumors (tumor penetration, overall required dose, nonuniform dose distributions owing to antigen tissue heterogeneity), it would appear that bringing RIT into a frontline therapeutic setting after induction chemotherapy and maximum cytoreduction would be the next logical direction. A phase III first-line indolent trial (FIT) of consolidation with Zevalin compared to no additional therapy after first remission was reported for follicular B-cell NHL.55 Patients with CD20+ stage III/IV follicular B-cell NHL who achieved a PR or CR to induction chemotherapy were randomized to Zevalin (n = 208) or to the control arm, representing no further treatment (n = 206). Prior to chemotherapy, patients had documented <25% bone marrow involvement. After induction chemotherapy, blood counts had to recover such that the ANC was ≥1.5, platelets were ≥150,000, and hemoglobin was ≥9. Patients in the Zevalin arm were treated with an activity of 0.4 mCi/kg; a maximum activity of 32 mCi was allowed. Although two doses of rituximab (250 mg/m2) were used, an 111In biodistribution scan was not required. The data was analyzed with a median follow-up of 3.5 years. Zevalin consolidation resulted in a median progression-free survival (PFS) advantage of 36.5 versus 13.3 months in the control arm (p <0.0001). The PFS benefit was maintained in the Zevalin arm regardless if patients achieved a PR (29.3 vs. 6.2 months; p <0.0001) or CR (53.9 vs. 29.5 months; p = 0.0154). The benefit of Zevalin consolidation was maintained across all Follicular Lymphoma International Prognostic Index (FLIPI) subgroups. In patients with a PR after induction chemotherapy, 77% were further converted to a CR when treated with Zevalin. This resulted in a final CR rate of 87% in the treatment arm, and this result compares well with established data. In the treatment arm, 90% of patients who were Bcl-2 positive converted to a negative status (90% molecular CR). Toxicity was well managed and primarily hematologic. A total of 8% of patients experienced a grade 3 to grade 4 infection. There were no treatment-related deaths. Data from the FIT trial have been updated.56 With a median follow-up of 66.2 months, the PFS advantage of Zevalin was maintained in patients undergoing a PR/CR. The overall survival was 93% and 89% for Zevalin versus the control arm, respectively (p = 0.561).
Results of a phase III randomized trial (S0016) was recently reported in abstract form.57 This study randomized 532 patients with bulky stage II through stage IV follicular NHL to cyclophosphamide, doxorubicin, vincristine, and prednisone (CHOP) regimens, specifically CHOP-R (n = 267) or CHOP-RIT (n = 265). The RIT consisted of a single consolidation dose of 131I-tositumomab, calculated to deliver 75 cGy to the total body; the chemotherapy consisted of six cycles of CHOP. In the CHOP-R arm, Rituximab was given on days 1, 6, 48, 90, 134, and 141. With a median follow-up of 4.9 years, the 2-year estimated PFS was 76% and 80% in the CHOP-R and the CHOP-RIT arms, respectively (p = 0.11). The overall survival was similar in both arms (p = 0.08).
 SOLID TUMOR TRIALS AND APPROVED THERAPEUTICS
SOLID TUMOR TRIALS AND APPROVED THERAPEUTICS
Lung cancer remains the leading cause of cancer mortality in the world. Clearly, new strategies are required to help improve local and systemic control. To date, most RIT-targeting constructs will bind to cell surface or extracellular matrix antigens, on or in surrounding viable malignant cells. Each antigenic target (disease site) requires a specific targeting construct (typically an mAb or antibody fragment). There is, however, emerging data to support the concept of targeting necrotic and hypoxic regions of tumors.58 The selective targeting of dead or dying cells will allow a cytotoxic event of nearby malignant cells by the bystander and crossfire effect. Additionally, only one type of targeting construct needs to be manufactured to target many different types of malignancies. If the cell surface antigen does not internalize to any significant degree, then a typical targeting construct will remain on the cell surface. Because dead and dying cells (undergoing apoptosis) exhibit disruption of their cell membrane, constructs that target intracellular products of apoptosis will then be able to gain access the cells’ cytoplasm and nucleus. Although several “dead cancer cell antigens” are under investigation, tumor necrosis therapy or treatment (TNT) has been investigated in human trials. TNT is an IgG2a mAb that targets nuclear histones.59–61
A pivotal trial of iodine-131-chimeric tumor necrosis treatment (131I-chTNT) in advanced lung cancer patients was performed.62 A total of 107 patients (n = 97, non–small cell; n = 10, small cell) were enrolled from 1999 to 2002. All patients had failed at least one prior therapeutic regimen (mean = 3; range = 1 to 5), and 86.9% of the patients had stage III to stage IV disease at study entry. In all cases, the patients received two instillations of 131I-chTNT administered over 2 to 4 weeks. Sixty-two patients received IV administrations, and 45 patients received intratumoral injections of 131I-chTNT. IV administrations were delivered at an activity 0.8 mCi/kg and intratumoral injections were delivered at an activity of 0.8 mCi/cm3 of tumor size. In all patients (n = 107), the ORR was 34.6% (3.7% CR; 30.8% PR; 55.1% no change or stable disease; 10.3% progressive disease). Of the 62 patients receiving a systemic administration of 131I-chTNT, the ORR was 35.5% (3.2% CR; 32.2% PR). Of the 45 patients receiving intratumoral injection of 131I-chTNT, the ORR was 33.3% (5% CR; 20.9% PR). In 58 evaluable patients, the median survival was 11.7 months, and the 1-year survival rate was 41.4%. The average absorbed doses for tumor and normal lung were 8.45 Gy and 2.35 Gy for patients receiving systemic 131I-chTNT and 30.0 Gy and 2.65 Gy for patients receiving intratumoral 131I-chTNT. The major toxicity was hematologic and reversible. As expected, the hematologic toxicity was lower in the intratumoral injection group. In 2003, 131I-chTNT was approved by the Chinese State Food and Drug Administration to treat refractory bronchogenic carcinoma. As a result, 131I-chTNT became the first solid tumor TRIT agent in the world approved for therapy. Recently, two studies using 131I-chTNT-1/B were completed for the treatment of CNS malignancies. The first trial was a phase I study using 131I-chTNT-1/B to treat progressive and recurrent glioblastoma multiforme (NCT00128635). The second trial was a phase II study using 131I-chTNT-1/B to treat patients with glioblastoma or anaplastic astrocytoma after conventional surgery.
HCC, or liver carcinoma, represents a significant worldwide malignancy and has several potential etiologies: viral, metabolic (hemochromatosis, alcoholic cirrhosis), toxins (aflatoxin), hormonal, or chemical (oil industry). In addition to resection, orthotopic liver transplantation (OLT) represents the only other potential curative option. In 1989, the Radiation Therapy Oncology Group (RTOG) reported a phase III study comparing EBRT and chemotherapy to the same treatment plus 131I antiferritin antibody. None of the patients receiving EBRT and chemotherapy only were converted to a resectable state. In a separate analysis, 11 patients crossing over from the EBRT and chemotherapy arm to further therapy with 131I antiferritin antibody were converted to resection. There was, however, no significant difference in the initial “intent to treat” treatment arms based on response rate and survival.63–64,65,66 Of course, the most promising role of radiolabeled antibody therapy is in the treatment of minimal residual microscopic disease.
Licartin is an antibody fragment, F(ab’)2, that targets HAb18G/CD147, a HCC TAA. More recently, the safety and pharmacokinetics of Licartin (131I metuximab) were investigated in phase I/II trials. The initial phase I trial evaluated 28 patients with HCC. They were treated with 0.25 to 1.0 mCi/kg of Licartin via hepatic artery infusion. In a subsequent phase II trial, 106 patients with HCC received 0.75 mCi/kg of Licartin on day 1 of a 28-day cycle. Life-threatening toxicities did not occur. In 73 patients (completing two cycles), 6 patients (8.22%) exhibited a PR, 14 patients (19.18%) had a minor response, and 43 patients (58.9%) maintained stable disease. The cohort had a 21-month survival of 44.54%. It was concluded that Licartin was safe and active in patients with HCC.67
Realizing that TRIT is most suited for treating microscopic disease, Licartin was tested in the adjuvant setting for patients with HCC undergoing OLT.64,68 A total of 60 patients with HCC who were undergoing OLT were randomized to Licartin (0.42 mCi/kg) for three fractions at 28-day intervals versus placebo. Analysis at 1 year post-therapy revealed that the recurrence rate was significantly decreased by 30.4% (p = 0.0174) and the survival rate was significantly increased by 20.6% (p = 0.0289) in the Licartin group. No significant toxicities were observed. The Chinese State Food and Drug Administration has approved Licartin as adjuvant therapy after OLT for HCC in 2005. There are two trials using Licartin that are ongoing but not recruiting patients. The first trial (NCT00819650) randomizes patients with HCC undergoing an R0 resection to postoperative Licartin or no further treatment. Licartin is delivered in three doses (fractions) at 28-day intervals beginning at week 4 after liver resection. In the second trial (NCT00829465), patients with unresectable HCC are randomized to transcatheter arterial chemoembolization (TACE) or Licartin combined with TACE.
Currently, the most common antigen targets for CNS malignancies consist of the epidermal growth factor receptor (EGFR), tenascin, neural cell adhesion molecule (NCAM), placental alkaline phosphatase (PLAP), and phosphatidyl inositide. The EGFR is variably amplified in malignant tissue and is also present, to some extent, in benign tissue. Tenascin is an extracellular glycoprotein that is uniformly expressed in glioma, and NCAM is present on both benign and malignant glioma cells. Clinical trials using RIT to treat CNS malignancies have been extensively reviewed.69 A phase III trial was reported in 2002.70 A total of 12 patients with malignant glioma were randomized to surgical resection and radiotherapy (60 Gy) (n = 5) versus surgical resection, radiotherapy, and RIT (n = 7). The RIT agent was a 125I-anti-EGFR antibody 425 that was administered intravenously in three weekly doses (50 mCi) beginning during week 4 of the EBRT. All patients in the treatment arm had a recurrence at the time of publication. Considering that the EGFR was not tested in submitted tissue, the trial had a small number of patients, and the 150-kDa antibody was administered intravenously, significant conclusions could not be drawn.
Most of the CNS RIT trials to date are of “dose searching pilot” or phase I design. The evolution of the trials has seen the delivery route move from systemic (intra-arterial or IV) to local instillation of the RIT agent into a surgically created resection cavity (SCRC). Even though the blood–brain barrier (BBB) is often disrupted by a rapidly growing CNS malignancy, this phenomenon is not well defined and 150 kDa antibodies would still not likely cross to a significant degree, although there does appear to be an element of nonspecific uptake from a systemic delivery.71 As a result, studies using the systemic approach often deliver EBRT in conjunction with TRT. It has been well documented that EBRT will cause an increase in the permeability of the BBB and increase vascular leakage.72–73,74 Regardless, it has been disappointingly estimated that only 0.001% to 0.01% of the systemically delivered antibody will penetrate each gram of solid tumor. Furthermore, biopsy data has revealed that a single systemic injection of radiolabeled anti-EGFR antibody will deliver only 0.02% of the injected activity per gram of tumor, resulting in a dose of only 100 to 200 cGy.71
Direct instillation of the TRT agent into the SCRC is an attractive alternative to the systemic approach. Unlike other malignant sites where the potential for systemic spread mandates a systemic approach, this is not the case for malignant gliomas. The local approach is accomplished by injecting or instilling the RIT agent directly into the SCRC via an Ommaya or Rickham catheter. Preliminary dosimetry is performed to ensure localization within the surgical bed and that no direct communication with the ventricular system has occurred. Institutions using this technique have utilized murine, chimeric, or humanized mAbs attached to 131I, 90Y, 188Re, and 211At. Other important treatment variances include fractionation, pretargeting, and a combined modality approach using EBRT and chemotherapy. The success of this approach will depend on meaningful penetration of the RIT agent into the local brain parenchyma such that the mAbs (or targeting construct) can bind to areas of microscopic extension of malignant cells at some distance from the SCRC margin. It is still unknown as to what impact the healing process/inflammation at the surgical margin has on the success of antibody penetration. As well, it is well known that binding site barrier phenomena, interstitial tumor pressure, aberrant tumor vasculature, and a recusant extracellular tumor matrix will significantly impede antibody penetration.75
Hopkins et al.76 obtained biopsy data from three patients with glioma who received two to three cycles of either 131I or 90Y-ERIC-1 (anti-NCAM antibody) directly instilled into a SCRC. Relevant assumptions were that the SCRCs were spherical, the radionuclide was spread evenly around the resection margin, 100% of the RIT agent was bound to its target, and diffusion into the resection margin was uniform. It was shown that “modest” diffusion occurred and the process was exponential. The peak dose occurred between 0.16 and 0.18 cm beyond the resection margin, and 4.4% to 5.8% of the peak dose was delivered to a depth of 2 cm. Of note, NCAM is expressed on benign and malignant cells and perhaps a more tumor-specific antigen would allow for greater depth of penetration. Certainly, smaller targeting constructs have been shown to penetrate to a greater depth in brain parenchyma compared to intact antibodies.77 Using the same antibody, radiolabeled with 131I and instilled into a SCRC, Papanatassiou73 showed that diffusion occurred from 0.5 to 1.0 cm (single-photon emission computed tomography [SPECT]). The range of antibody binding to the target was 8% to 80% of total injected activity. Because the R95 (thickness of tissue where 95% of the β energy is deposited) for 131I is only 0.992 mm, it was concluded that a more optimal radionuclide would potentially be 90Y with an R95 of 5.94 mm.78 Assuming a 2-cm SCRC and 100% binding, as much as 351 Gy could be delivered to the tumor with a single instillation of 18.2 mCi of 90Y-ERIC-1. This calculation resulted in an impressive minimum tumor/whole brain dose ratio of 140:1.
Using 131I-81C6 (antitenascin mAB), dose-limiting toxicity was reached with a single injection of 80 mCi for leptomeningeal disease (intrathecal delivery), 100 mCi for heavily pretreated and recurrent glioma (into SCRC), and 120 mCi for de novo glioma (into SCRC) also receiving EBRT and chemotherapy.79 Using a standard, fixed, mCi dose, a wide range of absorbed doses (18 to 186 Gy) will be delivered to a depth of 2 cm beyond the SCRC margin.80 On further analysis, an optimal dose of 44 Gy to 2 cm beyond SCRC was identified. Doses <44 Gy resulted in increased recurrence rates, and doses >44 Gy resulted in a higher rate of necrosis. A trend toward significant improvement in median survival was shown for patients receiving 40 to 48 Gy versus <40 Gy.78 Refining the technique further, it was shown that 20 of the 21 patients could be successfully dosed to 44 Gy by varying the initial injection activity and considering the volume of the SCRC.81 Zalutsky et al.82 showed that a high LET, α-emitting radioconjugate (211At-ch81C6) could be safely delivered in a small cohort of glioma patients. Interestingly, histopathology appears to correlate with prognosis. Biopsy data from patients with a suspected recurrence, after receiving 131I-labeled antitenascin 81C6 antibody, were analyzed. Three types of histologic patterns were evident: proliferative glioma, quiescent glioma, and negative for neoplasm. The median survival for each histopathologic pattern was 3.5, 15.0, and 27.5 months, respectively (p <0.0001). Considering total dose (EBRT plus radiolabeled antibody), patients receiving <86 Gy or >86 Gy had median survivals of 7 and 19 months, respectively (p <0.002).83
A review of the major RIT CNS trials69 indicates that the range of maximum tolerated activity is between 10 and 120 mCi. There are many variables that could potentially account for the noted range. In general, by performing dosimetry for a given radionuclide delivery construct, a specific absorbed dose can be calculated to a predetermined depth from the SCRC margin. It has been shown that 131I-antitenascin 81C6 can deliver 2,000 Gy, 90 Gy, and 34 Gy to the cavity interface at 1 cm and at 2 cm depth, respectively.78,83 The median survival for TRT in treating glioma appears extremely favorable when compared to other treatment approaches. For de novo lesions, the median survival range is 50.9 to 57.6 months (three studies not reaching median survival at the time of the report) for anaplastic astrocytoma and 13.4 to 35.5 months for glioblastoma. For recurrent lesions, the median survival range is 13.0 to 52.0 months (one study not reaching median survival at the time of the report) for anaplastic astrocytoma and 14.0 to 25.0 months for glioblastoma.69 Further improvements can be expected as this field matures and phase II and III data is generated. Unlike sealed source brachytherapy, there appears to be a very low rate of CNS toxicity and a reduced subsequent need for surgical intervention to remove necrotic regions.
Building on the data generated by Duke University, Bradmer Pharmaceuticals has developed two clinical trials using the 131I-antitenascin antibody (Neuradiab) for treating glioblastoma (World Health Organization [WHO] grade IV astrocytoma). The first trial (Glass-Art) is a phase III study that randomizes patients with untreated glioblastoma to standard therapy (surgery, radiotherapy, and temozolomide) versus standard therapy plus Neuradiab. In the experimental arm, dosimetry is performed and a calculated dose of radiolabeled antibody is delivered via a Rickham catheter. The Neuradiab therapeutic dose is given after surgery and prior to the initiation of radiotherapy and temozolomide. The second trial is a phase II study designed to treat patients with recurrent glioblastoma with surgery, Neuradiab, and bevacizumab. This trial is not yet open for patient recruitment. The Glass-Art trial began enrolling patients in 2008. Although “ongoing,” the trial is not recruiting participants at this time. Bradmer Pharmaceuticals continues to seek development partners and has submitted multiple grants for potential funding. On March 3, 2010, the company submitted a proposal for private placement. Funding continues to be problematic.
Initial promising investigations evaluated the long-term survival of patients with advanced ovarian cancer treated with RIT following cytoreductive surgery and platinum-based chemotherapy.84 Eligibility criteria included patients with histologic evidence of ovarian cancer from stage IC to stage IV. Fifty-two patients entered the study: 31 patients had residual disease following standard chemotherapy, and 21 patients had achieved CR. The treatment consisted of a single intraperitoneal (IP) administration of 25 mg HMFG1 labeled with 666 MBq (18 mCi)/m2 of 90Y, with survival being the primary end point. In the group of 21 patients who had achieved CR following surgery, conventional chemotherapy, and IP RIT, the median survival had not been reached with a maximum follow-up of 12 years. Survival at >10 years was 78%. The conclusion of this study was that a substantial proportion of patients who achieve a CR with conventional therapy can achieve a long-term survival benefit if treated with IP 90Y-HMFG1.
A phase III study was subsequently performed.85 This multinational (74 centers, 17 countries, recruiting patients between 1998 and 2003), open-label, randomized phase III study compared 90Y-HMFG1 (against the MUC 1 antigen) plus standard treatment versus standard treatment alone in patients with epithelial ovarian cancer (EOC) who had attained a complete clinical remission after cytoreductive surgery and platinum-based chemotherapy. Stage IC to stage IV patients were screened (n = 844), of whom 447 with a negative second-look laparoscopy (SLL) were randomly assigned to receive either a single dose of 90Y-HMFG1 plus standard treatment (224 patients) or standard treatment alone (223 patients). Patients in the active treatment (RIT) arm received an IP dose of 25 mg 90Y-HMFG1 to provide 666 MBq (18 mCi)/m2. After a median follow-up of 3.5 years, 70 patients had died in the active treatment arm compared with 61 patients in the control arm. Cox proportional hazards analysis of survival demonstrated no difference between treatment arms. In the RIT arm, 104 patients experienced relapse compared with 98 patients in the standard treatment arm. No difference in time to relapse was observed between the two study arms. The conclusion was that a single IP administration of 90Y-HMFG1 to patients with EOC, who had a negative SLL after primary therapy, did not extend survival or time to relapse. The reason for failure of the treatment could perhaps be explained by the choice of radionuclide. When treating microscopic disease with high-energy β-particles emitted from 90Y, the electron will have too long of a range to deliver high enough energy to the tumor cell nuclei. It has been extensively modeled that high-energy β-particle emissions will not deposit large amounts of energy (absorbed energy) into tumor spheroids below a certain size. However, there are other concerns about this study that warrant comment:
1. Entry was allowed onto the study even if dispersal of the TRT agent (90Y-HMFG1) could not occur in an entire quadrant of the abdomen because of adhesions. The adhesions were assessed by laparoscopy, computed tomography (CT) scan, or isotope diffusion scan. Although it is rather intuitive that a diffusion scan will allow a reasonable assessment of adhesions, it is less obvious that laparoscopy or a CT scan will discern between one or two quadrants of adhesions. This allowance could potentially result in a significant underdosing ≤25% of the abdominal cavity. Importantly, there was no mention of how these patients (no adhesions vs. one quadrant with adhesions) were stratified between each treatment arm.
2. The RIT arm contained 3% more stage III/IV patients.
3. The RIT arm had a higher mean CA-125 level after laparoscopy (never explained).
4. The RIT arm had 8% more patients with residual disease after initial surgery (44.2% vs. 35.9%).
5. In the standard arm, 7% (19.7% vs. 12.5%) more patients received consolidation chemotherapy.
6. The overall antibody mass (25 mg) may have been insufficient to help provide a concentration gradient to help “push” the radiolabeled antibody into the tumors.52 This can be contrasted to the 250 mg/m2 of cold antibody used with Zevalin or the 450-mg total antibody dose used with Bexxar.
7. With regard to the injected dose, 20% can enter the systemic circulation.
8. The radiolabeling process was performed by each institution and not centralized. Although a radiolabeling efficiency of 95% was confirmed with thin-layer chromatography, there was no mention of immunoreactivity quality assurance (potential loss of affinity of the antibody for the antigen as a result of the radiolabeling process).
9. During the accrual time period, single-institution experiences tended to report higher conversion rates to an open laparotomy (from an SLL) than was reported in the current study (59% vs. 5%).85
10. In the RIT arm, 18% of patients had ≤60% MUC1 staining (unknown impact).
11. There was no pattern of failure analysis (IP vs. distant).
The patterns-of-failure analysis eventually came to fruition.86 Case report forms of all patients with disease recurrence were reviewed to determine site and date of recurrent disease. The 447 patients were included with a median follow-up of 3.5 years. Relapse was seen in 104 of 224 patients in the RIT arm and 98 of 223 patients in the control arm. Significantly fewer IP (p <0.05) and more extraperitoneal (p <0.05) relapses occurred in the RIT arm. Time to IP recurrence was significantly longer (p = 0.0019) and time to extraperitoneal recurrence was significantly shorter for the RIT arm (p <0.001). In a subset analysis, the impact of IP RIT on IP relapse-free survival was even greater and could only be seen in a subgroup of patients with residual disease after primary surgery. Although there was no survival benefit for 90Y-HMFG1 IP instillation as consolidation treatment for EOC, an improved control of IP disease was found, which appeared to be offset by increased extraperitoneal recurrences. It was proposed that the transient myelosuppression (alteration of the immune system) induced by therapy with 90Y-HMFG1 indirectly caused the greater number of extraperitoneal metastases. Most likely, this observation is simply the result of an alteration in the failure pattern owing to a greater number of patients in the treatment arm benefiting from a greater IP control, as distant metastases will not be observed because of overwhelming local symptoms. In addition, for reasons mentioned previously, it is possible that the treatment arm was skewed with more advanced disease. Future trials should focus on both the IP and systemic delivery of RIT.
 UNSEALED RADIONUCLIDE THERAPY
UNSEALED RADIONUCLIDE THERAPY
Unsealed radionuclide therapy (URT) refers to the medical application of radiopharmaceuticals that are not conjugated to a targeting agent and thereby localize in diseased tissue by virtue of biologic, chemical, or physical avidity.87 These radionuclides are considered “unsealed” because they are not confined within a container that could be inserted or implanted into a tumor, as is performed with conventional brachytherapy techniques. Because they are not conjugated to a traditional targeting construct, this class of therapeutics has also been referred to as “naked” radiopharmaceuticals.88 Oversight for the utilization of URT is governed by the U.S. FDA.89 Safety issues, radioactive material shipping, and licensing are regulated by the U.S. Nuclear Regulatory Commission (NRC). A state may enter into an agreement with the NRC to perform its own regulation and to monitor of the use of radioactive material (agreement state), with the exception of fuel facilities and nuclear reactors. States that continue to allow monitoring by the NRC are referred to as nonagreement states.90 The NRC receives advice regarding radiopharmaceuticals from the Advisory Committee on the Medical Use of Isotopes (ACMUI). Regulations for the practice of nuclear medicine reside in U.S. NRC Title 10 of the Code of Federal Regulations, Parts 20 and 35. Part 20 largely governs the standards for radiation protection, and Part 35 governs the medical use of radioactive material.91,92
Bone-seeking radiopharmaceuticals, used for palliation of painful bone metastases, represent one of the more common uses of URT. A few of the earlier radionuclides used for this purpose include phosphorus-32 [32P], samarium-153 [153Sm], and strontium-89 [89Sr];93 however, newer agents are being investigated and are in various stages of development.94–95,96–101 As with RIT, the radionuclides used in the application of URT can be classified as b-, α-, and Auger emitters. The radionuclides used in URT target bone by either an intrinsic affinity (i.e., 89Sr, radium-223 [223Ra]) or by using bone-seeking phosphonate ligands attached to the radionuclide (i.e., samarium-153 ethylenediaminetetramethylenephosphonate [153Sm-EDTMP] or rhenium-188 hydroxyethylidine diphosphate [188Re-HEDP]).95,102 Localization properties of individual agents and the clinical circumstances involved will determine routes of administration. These agents have been delivered by IV, intra-arterial, intracavitary, intra-articular (radiosynovectomy), and direct intralesional approaches. This variability of administration has been especially true for 32P, which has been uniquely studied using IV, oral, IP, and intrathoracic routes.103
The initial use of a b-emitting radioisotope for the management of intractable malignant bone pain was reported in 1942.104 Because URT demonstrates a chemical affinity for bone, the predominant thrust of clinical investigation has been for primary and secondary malignancies of bone and bone marrow. Other target sites, however, have been considered. In some instances, these alternative uses have remained a part of the therapeutic armamentarium; however, for many indications, the use of URT has yielded to nonradioactive approaches (corticosteroids, systemic chemotherapy, hormone therapy, analgesia, and surgery) and to EBRT. The lack of access to innovative candidate radionuclides, diminished trial participation, and absence of utilization and teaching from many training programs have further exacerbated the problem.87 Regardless, a review of 15 randomized controlled trials (1,146 analyzed patients) comparing URT to placebo or another radionuclide for the treatment of metastatic bone pain confirms the efficacy of URT for pain management. This review also provides evidence that URT resulted in significant and complete pain relief during a 1- to 6-month period.105 Practice guidelines have been established for URT,106 and evidence-based guidelines for palliation of bone metastases include URT as a reasonable therapeutic option.107
The most common malignant sites that develop bone metastases are prostate, breast, and lung cancer.93 URT exhibits increased targeting of bone in areas of osteoblastic activity and exerts this propensity because of a chemical similarity to calcium, which is classified as an alkaline earth metal in the periodic table (as are 89Sr and 223Ra). These therapeutic agents may either directly substitute for stable analogues in hydroxyapatite or may be chemisorbed on the hydroxyapatite surface of the phosphate moiety of phosphonate chelates.102 Radionuclide decay profiles that include gamma emissions may be utilized for imaging and for documentation of therapeutic uptake in regions of bone pathology. Bone scans (technetium-99 m [99mTc]) are typically performed to verify disseminated osseous disease. The intensity of uptake on pretherapeutic scanning does not necessarily coincide with therapeutic efficacy, and widely disseminated disease may actually produce “dilution” of dose and potentially reduced effectiveness.102
Regardless of the precise method of chemical or physical affinity, all agents studied for palliation of osseous metastatic bone pain have fared better than placebos in randomized trials.107 There is, however, limited evidence that the response or morbidity profile of the different radiopharmaceuticals vary significantly among themselves. Additionally, there is little evidence that dose escalation either in individual or cumulative doses will improve effectiveness. Sequenced administration has been investigated; however, if an initial intervention has not produced a significant level or duration of response, there is little evidence that additional administrations will increase effectiveness but they may potentially increase morbidity.102
Rapid and significant localization in bone by all agents will generally limit potential morbidity to myelosuppression, which in patients with adequate marrow reserve will usually be mild and be manifest initially within 1 week post-administration with evidence of thrombocytopenia. Leukopenia may develop somewhat later; however, all side effects typically reverse without intervention within 8 to 10 weeks. Circulating isotope not immediately incorporated into bone is typically excreted in urine; therefore, patients with reduced renal function may not be ideal candidates for the agents and, if used, should have blood counts monitored carefully. Administration is routinely on an outpatient basis, thus radiation protection measures for low-level radiation in urine should be practiced.90
Palliative effects may be observed within 3 to 5 days but usually peak at approximately 7 to 10 days, and the beneficial effects may last for months. At this point in time, subsequent administrations may be considered. When used for management of bone pain, all bone-seeking radiopharmaceuticals can exhibit a flare in pain within 24 to 72 hours post-injection that may last for 5 to 7 days. Appropriate analgesic management must be provided during this period. In the treatment of metastatic prostate cancer, prostate-specific antigen (PSA) levels may begin to decline within several days; however, the rapidity of decline, nadir of the PSA level, and duration of PSA response are not satisfactory predictors of improved outcomes.102
The bone-seeking agents have been and continue to be used primarily in metastatic prostate cancer, and evidence of effectiveness in breast and lung cancer is limited with responses noted primarily in osteoblastic metastases. Plain radiographs of symptomatic metastatic sites should be obtained prior to the use of systemic agents for palliation of bone pain. If there is evidence of possible impending fracture, stabilization and/or EBRT should be initiated prior to systemic radionuclide therapy. If painful vertebral metastasis is apparent clinically, CT or magnetic resonance imaging (MRI) of the painful vertebral segments should be obtained prior to administration of isotope to ensure that no epidural disease is present. If this pathology is discovered, EBRT or surgery should be carried out prior to systemic isotope therapy. IV administration of all agents should be carried out slowly over 1 to 5 minutes, through indwelling catheters with clear and unobstructed flow clearly validated, adequate hydration, and careful radiation precautions for patients and staff.
Radiopharmaceuticals
Iodine-131
Physical Properties: t½ = 8.0 days; radiation decay: β (606 keV maximum and 190 keV mean); γ (364 keV).
Clinical Utility: Radioiodine (131I) was first used to treat benign thyroid disease in the late 1920s and early 1930s.108,109 The first reports of using 131I to treat well-differentiated thyroid cancer (WDTC) were published in the 1940s.110,111 To date, 131I has become the standard of care, in conjunction with surgery, for the management of WDTC; its use and indications have been extensively reviewed.110,112,113 Benign thyroid tissue (follicular cells) and certain thyroid carcinomas (follicular, papillary, and Hürthle cell carcinoma) will actively transport iodine into the cell via the sodium iodide symporter to initiate the synthesis thyroid hormone.114,115 As a result, this innate targeting system has been exploited for many decades to treat locally persistent, recurrent, or metastatic thyroid carcinoma. The majority of data concerning the treatment of WDTC with 131I has been generated by large retrospective series of patients, with the resulting clinical data often spanning several decades. Frequently, these institutions used unchanged protocols and fixed activities for therapy. Regardless, considerable evidence exists concerning local control, decreased metastases, and a survival benefit when 131I is used as part of the treatment regimen.116,117–118,119
In general, 131I therapy for WDTC is considered either ablation or treatment. Ablation is the use of 131I to sterilize normal remnant thyroid tissue or microscopic disease that remains after thyroidectomy. Treatment refers to the therapeutic application of 131I against cancer persistence, local recurrence, or distant metastatic disease. Whereas the treatment aspect of 131I is well accepted, ablation is more controversial.120,121 Recent decades have revealed a decrease in mortality for WDTC, owing to the early diagnosis and aggressive treatment of WDTC with near-total thyroidectomy and 131I ablation in selected patients at high risk for recurrence and mortality, followed by thyroid-stimulating hormone (TSH) suppression.122 If it is determined that ablation will be performed, patients are placed on a low-iodine diet and TSH stimulation is performed by withholding thyroid hormone.113 Standard fixed activities of 30 to 100 mCi are used for ablation, whereas higher activities in the range of 100 to 300 mCi are used for known residual disease, recurrence, or metastatic disease. If possible, patient-specific dosimetry should be used to determine the activity in the metastatic setting.122 Using this approach, a dose <200 cGy is calculated to the blood (bone marrow) and ≤120 mCi of 131I being retained after 48 hours. This can result in administered activities between 75 and 659 mCi without the development of leukemia, permanent bone marrow suppression, or pulmonary fibrosis.123 Diagnostic 131I scanning and thyroglobulin measurement are required for follow-up.
Phosphorus-32
Physical Properties: t½ = 14.3 days; radiation decay: β (1.71 MeV maximum and 1.69 MeV mean); γ (none).
Clinical Utility: Phosphorus-32 (32P) represents one of the earliest agents in this class and perhaps is the most frequently studied for a wide variety of indications and routes of administration. In its aqueous form (Na2PO3), the agent was employed for the systemic therapy of chronic myelogenous leukemia and polycythemia vera. Orthopedic surgeons and rheumatologists have evaluated the agent for intra-articular management of persistent synovial effusions and hemarthroses secondary to hemophilia and leukemias.124,125 The agent has been placed in indwelling catheters to treat CNS lesions.
The colloidal form of the agent (as chromic phosphate) became a standard modality for management of malignant pleural and peritoneal effusions in the 1960s and 1970s, driving numerous clinical investigations. Anecdotal reports suggesting significant activity were infrequently corroborated in randomized clinical trials. Following drainage of abdominal ascites or pleural effusions, up to 5 mCi of the agent was instilled and patients were placed in various positions to enhance distribution. The nature of the disease processes and prior therapy often predisposed patients to preinstillation adhesions with bowel or lung immobility, and distribution of the agent proved difficult. This indication has largely been replaced by instillations of various antibiotic or chemotherapeutic compounds.126,127–128
Following identification of a subset of early-stage, high-risk ovarian cancer patients (FIGO stage Ia or Ib [grade 3], or stage 1c or II [any grade], or any stage I/II patient with clear-cell histology), the Gynecologic Oncology Group (GOG), North Central Cancer Treatment Group (NCCTG), and Southwest Oncology Group (SWOG) undertook a randomized trial assigning patients to either a single dose of 15 mCi of IP 32P versus cyclophosphamide 1 g/m2 and cisplatin 100 mg/m2 every 21 days for three cycles. Prior to instillation of the radioactive material through multiperforated indwelling peritoneal dialysis catheters, 99mTc was instilled to ensure free-flow and even distribution of the therapeutic agent. IP 32P was administered within 10 days but not >6 weeks following laparotomy. Ten-year follow-up of the study population suggested a modest reduction in intra-abdominal recurrence rate for the chemotherapy population but only a small and nonsignificant improvement in survival. These findings were corroborated by other reports.129
In the 1990s, Order et al.130 reported a series of patients treated with direct intralesional infusions of 32P colloidal chromic phosphate for unresectable tumors of the liver, CNS, pancreas, and head and neck. The observation of extralesional extravasation of isotope was apparently overcome by preinstillation of macroaggregated albumin to induce capillary and arteriole blockade prior to isotope infusion. Intense activity and doses were documented; however, improvements in local control and survival were inconclusive.131
32P localization in bone created interest in use of the orthophosphate form of the isotope for painful skeletal metastases with 85% of the administered dose ultimately incorporated into bone. However, priming regimens including androgenic agents prior to isotope administration were prolonged, beneficial results modest, and myelotoxicity significant; use of the agent for this indication has largely been abandoned.
Strontium-89 Chloride (Metastron, GE Healthcare, Chalfont St. Giles, UK)
Physical Properties: t½ = 50.5 days; radiation decay: β (1.463 MeV maximum and 0.583 MeV mean); γ (none).
89Sr, a calcium analogue, is administered as an IV injection at doses of 4 mCi, given slowly.
Clinical Utility: Porter et al.,132 in a Trans-Canada study, compared 89Sr to bisphosphonates in the prophylactic setting in an attempt to reduce subsequent development of additional osseous metastasis. Results were comparable in both arms, with a reduced cost of therapy in the 89Sr arm. Low-grade hematologic toxicity in the radiation arm did not require intervention. In an attempt to build on currently established palliative results, a randomized phase III trial is under way to investigate the use of weekly doxorubicin (20 mg/m2) with 89Sr after response to induction chemotherapy.133
Samarium-153 Lexidronam (Quadramet, Cytogen, Princeton, NJ)
Physical Properties: t½ = 46.3 hours; radiation decay: β (0.81 MeV maximum and 0.23 MeV mean); γ (maximum energy 103 keV).
Clinical Utility: Samarium-153 EDTMP (153Sm) is a bone-seeking agent consisting of radioactive samarium and a telephosphonate chelator, EDTMP. The recommended therapeutic dose is 1.0 mCi/kg, administered intravenously over a period of 1 minute through a secure indwelling catheter and followed by a saline flush. Extensive preclinical and clinical investigations have demonstrated the safety and effectiveness profile of 153Sm-EDTMP.134 Although primarily used alone, there is increasing interest in consideration of combination therapy with the bisphosphonates and taxane-based chemotherapeutics.
The use of 153Sm-EDTMP has been evaluated in osseous metastases for primary osteosarcomas. Anderson et al.135 investigated the use of gemcitabine as a radiosensitizer to increase 153Sm-EDTMP effectiveness. Using 30 mCi/kg (average of 1,640 mCi), they found acceptable toxicity and objective response in 8 of 14 patients investigated.
Radium-223 Chloride
Physical Properties: t½ = 11.4 days; radiation decay: α (6 MeV maximum); γ (270 keV maximum).
Clinical Utility: Although not available for commercial distribution in the United States, there has been interest in 223Ra in the treatment of metastatic hormone-refractory prostate cancer involving multiple bones. Nilsson et al.136 at the Karolinska University Hospital and Institute in Stockholm reported initial findings in 2005, evaluating the safety and effectiveness of 223Ra. The group had carried out preclinical studies prior to this phase I investigation. They concluded that at relevant dose levels, the agent was well tolerated and justified extension into phase II and III studies. Following focal EBRT to selected sites, patients were randomly assigned to treatment with 223Ra or placebo in a double-blinded manner. One primary end point—bone-alkaline phosphatase levels—was significantly decreased in the treatment arm, and the time to PSA increase was significantly lengthened. Hematologic toxicity in both groups was equivalent. The time to first skeletal-related event (SRE) was significantly increased in the 223Ra cohort and can be considered as a measure of quality of life, taking into account increase in pain or analgesic requirements; new neurologic symptoms or fractures; or additional surgical, radiologic, or systemic therapy. In this limited report, survival in the treated group was significantly extended. Subsequent reports have confirmed these findings, and a commercially prepared formulation of the agent (Alpharadin, Algeta ASA, Oslo, Norway) was granted approval by the U.S. FDA in February 2008 for phase I/II trials in the United States.137 Currently, U.S. patients are eligible for ALSYMPCA (Alpharadin in Symptomatic Prostate Cancer), an international phase III trial randomizing patients with hormone-refractory prostate cancer to Alpharadin plus best standard of care versus placebo plus best standard of care.
Rhenium-186 HEDP (Etidronate)
Physical Properties: t½ = 3.8 days; radiation decay: β (1.07 MeV maximum and 0.336 mean); γ (0.137 MeV maximum).
Clinical Utility: Although not commercially available in the United States, 186Rh-HEDP has been studied in phase I trials in Europe in association with autologous peripheral blood stem cell rescue in the management of hormone-refractory prostate cancer metastatic to bone.138 Promising results have led to initiation of phase II trials.
Rhenium-188 HEDP (Etidronate)
Physical Properties: t½ = 16.9 hours; radiation decay: β (2.1 MeV maximum and 0.779 mean); γ (0.155 MeV maximum and 0.061 mean).
Clinical Utility: Although not commercially available in the United States, 188Re-HEDP has been studied in Europe for some time. The agent is produced by a generator similar to that used to produce 99mTc, enabling wide availability at relatively low cost. Liepe et al.139 reported treatment of 46 patients with multiple bone metastases from breast and prostate cancer with pain. Thirty-one patients received 188Re-HEDP (3,300 MBq) and 25 patients received 153Sm-EDTMP (37 MBq/kg of body weight). All patients had a single injection of isotope. Patients with prostate cancer received hormone therapy for 6 months before isotope therapy and during the post-isotope observation period. Thirty-nine patients received bisphosphonates for 6 months prior to study treatment with discontinuance of the agents 1 month prior to isotope administration. In post-therapy evaluation, only the 188Re-HEDP group had a statistically significant improvement in the Karnofsky Performance score. Pain relief within 2 weeks of treatment was noted in 77% of the 188Re-HEDP group and in 73% of the 153Sm-EDTMP group. These results were not statistically significant, and there was no significant difference between responses in the patients with prostate or breast cancer. A brief flare reaction was noted in 17% of patients in both groups within 14 days of therapy, and the majority of patients demonstrated a maximum of grade I anemia within 12 weeks of therapy based on the 1979 WHO criteria. Grade I thrombocytopenia was noted in 2 patients with each isotope, and 1 patient in the 188Re-HEDP group experienced grade II thrombocytopenia. Grade I leukopenia was noted in 1 patient in each group. All cases of thrombocytopenia and leukopenia reversed within 12 weeks after therapy. Similar findings have been reported by other investigators.140,141
 REFERENCES
REFERENCES
1. Abbas AK, Lichtman AH, Pillais S, eds. Cellular and molecular immunology. Philadelphia: Saunders Elsevier, 2007.
2. Campoli M, Ferrone S. Cancer immune surveillance and tumor escape mechanisms. In: Speer TW, ed. Targeted radionuclide therapy. Philadelphia: Lippincott Williams & Wilkins, 2011:3–21.
3. Jeoung DI. Employing SEREX for identification of targets for anticancer targeted therapy. In: Speer TW, ed. Targeted radionuclide therapy. Philadelphia: Lippincott Williams & Wilkins, 2011:159–167.
4. Wong JYC, Williams LE, Yazaki PJ. Radioimmunotherapy of colorectal cancer. In: Speer TW, ed. Targeted radionuclide therapy. Philadelphia: Lippincott Williams & Wilkins, 2011:321–351.
5. O’Donoghue JA. Dosimetric principles of targeted radiotherapy. In: Abrams PG, Fritzberg AR, eds. Radioimmunotherapy of cancer. New York: Marcel Dekker, 2000:1–20.
6. Burvenich IJG, Scott AM. The delivery construct: maximizing the therapeutic ratio of targeted radionuclide therapy. In: Speer TW, ed. Targeted radionuclide therapy. Philadelphia: Lippincott Williams & Wilkins, 2011:236–248.
7. DiCara D, Nissim A. Methods for development of monoclonal antibody therapeutics. In: Speer TW, ed. Targeted radionuclide therapy. Philadelphia: Lippincott Williams & Wilkins, 2011:22–31.
8. Kohler G, Milstein C. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature 1975;256(5517):495–497.
9. Jain M, Kaur S, Batra SK. Modulation of biologic impediments for radioimmunotherapy of solid tumors. In: Speer TW, ed. Targeted radionuclide therapy. Philadelphia: Lippincott Williams & Wilkins, 2011:182–190.
10. Carmeliet P, Jain RK. Angiogenesis in cancer and other diseases. Nature 2000;407:249–257.
11. Jain RK. Transport of molecules in the tumor interstitium: a review. Cancer Res 1987;47:3039–3051.
12. Padera TP, Kadambi A, di Tomaso E, et al. Lymphatic metastasis in the absence of functional intratumor lymphatics. Science 2002;296:1883–1886.
13. Zwanziger D, Beck-Sickinger AG. Malignancies treated with peptides. In: Speer TW, ed. Targeted radionuclide therapy. Philadelphia: Lippincott Williams & Wilkins, 2011:483–497.
14. Zwanziger D, Beck-Sickinger AG. Radiometal targeted tumor diagnosis and therapy with peptide hormones. Curr Pharm Des 2008;14:2385–2400.
15. Imhof A, Brunner P, Marincek N, et al. Response, survival, and long-term toxicity after therapy with the radiolabeled somatostatin analogue [90Y-DOTA]-TOC in metastasized neuroendocrine cancers. J Clin Oncol 2011;29:2416–2423.
16. Seregni E, Maccauro M, Coliva A, et al. Treatment with tandem [(90Y]DOTA-TATE and [(177)Lu] DOTA-TATE of neuroendocrine tumors refractory to conventional therapy: preliminary results. Q J Nucl Mol Imaging 2010;54:84–91.
17. Stahl S, Friedman M, Carlsson J, et al. Affibody molecules for targeted radionuclide therapy. In: Speer TW, ed. Targeted radionuclide therapy. Philadelphia: Lippincott Williams & Wilkins, 2011:49–58.
18. Friedman M, Stahl S. Engineered affinity proteins for tumour-targeting applications. Biotechnol Appl Biochem 2009;53:1–29.
19. Wallberg H, Ahlgrens S, Widstrom C, et al. Evaluation of the radiocobalt-labeled [MMA-DOTA-Cys(61)]-Z (HER2:2395)-Cys affibody molecule for targeting of HER2-expressing tumors. Mol Imaging Biol 2010;12:54–62.
20. Missailidis S, Perkins A. Radiolabeled aptamers for imaging and therapy. In: Speer TW, ed. Targeted radionuclide therapy. Philadelphia: Lippincott Williams & Wilkins, 2011:59–70.
21. Mayer G. The chemical biology of aptamers. Angew Chem Int Ed Engl 2009;48:2672–2689.
22. Goodchild J. Therapeutic oligonucleotides. Methods Mol Biol, 2011;764:1–15.
23. Hicke BJ, Stephens AW, Gould T, et al. Tumor targeting by an aptamer. J Nucl Med 2006;47:668–678.
24. Burvenich IJG, Scott AM. The delivery construct: maximizing the therapeutic ratio of targeted radionuclide therapy. In: Speer TW, ed. Targeted radionuclide therapy. Philadelphia: Lippincott Williams & Wilkins, 2011:236–248.
25. Murdy JS, Siegel RW, Stein J, et al. Translational nanomedicine: status assessment and opportunities. Nanomedicine 2009;5:251–273.
26. Mitra A, Nan A, Line BR, et al. Nanocarriers for nuclear imaging and radiotherapy of cancer. Curr Pharm Des 2006;12:4729–4749.
27. Chithrani BD, Stewart J, Allen C, et al. Intracellular uptake, transport, and processing of nanostructures in cancer cells. Nanomedicine 2009;5:118–127.
28. Maeda H, Sawa T, Konno T. Mechanism of tumor-targeted delivery of macromolecules drugs, including the EPR effect in solid tumor and clinical overview of the prototype polymeric drug SMANCS. J Control Release 2001;74:47–61.
29. Tesauro D, Morelli G, Pedone C, et al. Radiolabeled peptides, structure and analysis. In: Speer TW, ed. Targeted radionuclide therapy. Philadelphia: Lippincott Williams & Wilkins, 2011:32–48.
30. Pedone C, Morelli G, Tesauro D, et al. Peptide structure and analysis. In: Chinol M, Paganelli G, eds. Radionuclide peptide cancer therapy. New York: Taylor & Francis Group, 2006:1–30.
31. Fowler JF. Radiobiological aspects of low dose rate in radioimmunotherapy. Int J Radiat Oncol Biol Phys 1990;18:1261–1269.
32. Murry D, McEwan AJ. Radiobiology of systemic radiation therapy. Cancer Biother Radiopharm 2007;22:1–23.
33. Speer TW, Khuntia D. Introduction to radiation therapy. In: Mehta MP, ed. Principles and practice of neuro-oncology: a multidisciplinary approach. New York: Demos Medical Publishing, 2011:719–743.
34. Speer TW, Limmer JP, Henrich D, et al. Evolution of radiotherapy toward a more targeted approach for CNS malignancies. In: Speer TW, ed. Targeted radionuclide therapy. Philadelphia: Lippincott Williams & Wilkins, 2011:356–376.
35. Bernhardt P, Speer TW. Modeling the systemic cure with targeted radionuclide therapy. In: Speer TW, ed. Targeted radionuclide therapy. Philadelphia: Lippincott Williams & Wilkins, 2011:263–280.
36. Wilson AD, Brechbiel MW. Chelation chemistry. In: Speer TW, ed. Targeted radionuclide therapy. Philadelphia: Lippincott Williams & Wilkins, 2011:88–107.
37. Witzig TE. Radioimmunotherapy for B-cell non-Hodgkin lymphoma. In: Reilly RM, ed. Monoclonal antibody and peptide-targeted radiotherapy of cancer. Hoboken, NJ: John Wiley & Sons, 2010:169–218.
38. Reardan DT, Meares CF, Goodwin DA, et al. Antibodies against metal chelates. Nature 1985;316:265–268.
39. Le Doussal JM, Martin M, Gautherot E, et al. In vitro and in vivo targeting of radiolabeled monovalent and divalent haptens with dual specificity monoclonal antibody conjugates: enhanced divalent hapten affinity for cell-bound antibody conjugate. J Nucl Med 1989;30:1358–1366.
40. Boerman OC, Kranenborg MH, Oosterwijk E, et al. Pretargeting of renal cell carcinoma: improved tumor targeting with bivalent chelate. Cancer Res 1999;59:4400–4405.
41. Rossi EA, Sharkey RM, McBride W, et al. Development of new multivalent-bispecific agents for pretargeting tumor localization and therapy. Clin Cancer Res 2003;9:3886S–3896S.
42. Sharkey RM, Goldenberg DM. Pretargeted radioimmunotherapy. In: Speer TW, ed. Targeted radionuclide therapy. Philadelphia: Lippincott Williams & Wilkins, 2011:191–208.
43. Axworthy DB, Reno JM, Hylarides MD, et al. Cure of human carcinoma xenographs by a single dose of pretargeted yttrium-90 with negligible toxicity. Proc Natl Acad Sci 2000;97:1802–1807.
44. Sung S, Van Osdol WW. Pharmacokinetic comparison of direct antibody targeting with pretargeting protocols based on streptavidin-biotin binding. J Nucl Med 1995;36:867–876.
45. Sharkey RM, Goldenberg DM. Cancer radioimmunotherapy. Immunotherapy 2011;3:349–370.
46. Song H, Sgouros G. Radioimmunotherapy of solid tumors: searching for the right target. Curr Drug Deliv 2011;8:26–44.
47. Lin FI, Iagaru A. Current concepts and future directions in radioimmunotherapy. Curr Drug Discov Technol 2010;7:253–262.
48. Chatal JF, Davodeau F, Cherel M, et al. Different ways to improve the clinical effectiveness of radioimmunotherapy in solid tumors. J Cancer Res Ther 2009;5:S36–S40.
49. Boerman OC, Koppe MJ, Postema EJ, et al. Radionuclide therapy of cancer with radiolabeled antibodies. Anticancer Agents Med Chem 2007;7:335–343.
50. Burdick M, Macklis RM. Radioimmunotherapy for non-Hodgkin lymphoma: a clinical update. In: Speer TW, ed. Targeted radionuclide therapy. Philadelphia: Lippincott Williams & Wilkins, 2011:426–440.
51. Sharkey RM, Karacay H, Goldenberg DM. Improving the treatment of non-Hodgkin lymphoma with antibody-targeted radionuclides. Cancer 2010;116:1134–1145.
52. Pandit-Taskar N, O’Donoghue JA, Morris MJ, et al. Antibody mass escalation study in patients with castration-resistant prostate cancer using 111In-J591:lesion delectability and dosimetric projections for 90Y radioimmunotherapy. J Nucl Med 2008;49:1066–1074.
53. Witzig TE, Gordon LI, Cabanillas F, et al. Randomized, controlled trial of yttrium-90-labeled ibritumomab tiuxetan radioimmunotherapy versus rituximab immunotherapy for patients with relapsed refractory low-grade, follicular, or transformed B-cell non-Hodgkin’s lymphoma. J Clin Oncol 2002;20:2453–2463.
54. Kaminski MS, Zelenetz AD, Press OW, et al. Pivotal study of iodine I-131-tositumomab for chemotherapy-refractory, low-grade, or transformed low-grade B-cell non-Hodgkin’s lymphomas. J Clin Oncol 2001;19:3918–3928.
55. Morschhauser F, Radford J, Van Hoof A, et al. Phase III trial of consolidation therapy with yttrium-90-ibritumomab tiuxetan compared with no additional therapy after first remission in advanced follicular lymphoma. J Clin Oncol 2008;26:5156–5164.
56. Hagenbeek A, Radford J, Hoof AV, et al. 90Y-ibritumomab tiuxetan (Zevalin) consolidation of first remission in advanced-stage follicular non-Hodgkin’s lymphoma: updated results after a median follow-up of 66.2 months from the international, randomized, phase III first-line indolent trial (FIT) in 414 patients [abstract 594]. 52nd ASH Annual Meeting and Exposition, December 6, 2010, Orlando, FL. Available at: http://ash.confex.com/ash/2010/webprogram/Paper28386.html. Accessed January 1, 2012.
57. Press OW, Unger JM, Rimsza LM, et al. A phase III randomized intergroup trial (SWOG S0016) of CHOP chemotherapy plus rituximab Vs. CHOP plus iodine-131-tositumomab for the treatment of newly diagnosed follicular non-Hodgkin’s lymphoma [abstract 98]. 53rd ASH Annual Meeting and Exposition, December 11, 2011, San Diego, CA. Available at: http://ash.confex.com/ash/2011/webprogram/Paper39037.html. Accessed January 1, 2012.
58. Al-Ejeh F, Brown MP. Combined modality therapy: relevance for targeted radionuclide therapy. In: Speer TW, ed. Targeted radionuclide therapy. Philadelphia: Lippincott Williams & Wilkins, 2011:220–235.
59. Yu L, Ju DW, Chen W, et al. 131I-chTNT radioimmunotherapy of 43 patients with advanced lung cancer. Cancer Biother Radiopharm 2006;21:5–14.
60. Street HH, Goris ML, Fisher GA, et al. Phase I study of 131I-chimeric(ch) TNT-1/B monoclonal antibody for the treatment of advanced colon cancer. Cancer Biother Radiopharm 2006;21:243–256.
61. Wang H, Cao C, Li B, et al. Immunogenicity of iodine 131 chimeric tumor necrosis therapy monoclonal antibody in advanced lung cancer patients. Cancer Immunol Immunother 2008;57:677–684.
62. Chen S, Yu L, Jiang C, et al. Pivotal study of iodine-131-labeled chimeric tumor necrosis treatment radioimmunotherapy in patients with advanced lung cancer. J Clin Oncol 2005;23:1538–1547.
63. Order SE, Stillwagon GB, Klein JL, et al. Iodine 131 antiferritin, a new treatment modality in hepatoma: a Radiation Therapy Oncology Group study. J Clin Oncol 1985;3:1573–1582.
64. Order S. Radioimmunotherapy of unresectable hepatocellular carcinoma. In: Speer TW, ed. Targeted radionuclide therapy. Philadelphia: Lippincott Williams & Wilkins, 2011:352–355.
65. Leichner PK, Klein JL, Siegelman SS, et al. Dosimetry of 131I-labeled antiferritin in hepatoma: specific activities in the tumor and liver. Cancer Treat Rep 1983;67:647–658.
66. Order SE, Pajak T, Leibel S, et al. A randomized prospective trial comparing full dose chemotherapy to 131I antiferritin: an RTOG study. Int J Rad Oncol Biol Phys 1989;20:953–963.
67. Chen Z-N, Mi L, Xu J, et al. Targeting radioimmunotherapy of hepatocellular carcinoma with iodine (131I) metuximab injection: clinical phase I/II trials. Int J Radiat Oncol Bio Phys 2006;65:435–444.
68. Xu J, Shen Z-Y, Chen X-G, et al. A randomized controlled trial of Licartin for preventing hepatoma recurrence after liver transplantation. Hepatology 2007;45:269–276.
69. Speer TW, Limmer JP, Henrich D, et al. Evolution of radiotherapy toward a more targeted approach for CNS malignancies. In: Speer TW, ed. Targeted radionuclide therapy. Philadelphia: Lippincott Williams & Wilkins, 2011:356–376.
70. Wygoda Z, Kula D, Bierzynska-Macyszyn G, et al. Use of monoclonal anti-EGFR antibody in the radioimmunotherapy of malignant gliomas in the context of EGFR expression in grade III and IV tumors. Hybridoma 2006;26(3):125–132.
71. Kalofonos HP, Pawlikowska TR, Hemingway A, et al. Antibody guided diagnosis and therapy of brain gliomas using radiolabeled monoclonal antibodies against epidermal growth factor receptor and placental alkaline phosphatase. J Nucl Med 1989;30:1636–1645.
72. Brady LW, Markoe AM, Woo DV, et al. Iodine-125-labeled anti-epidermal growth factor receptor-425 in the treatment of glioblastoma multiforme. Front Radiat Ther Oncol 1990;24:151–160; discussion 161–165.
73. Papanastassiou V, Pizer BL, Coakham HB, et al. Treatment of recurrent and cystic malignant gliomas by a single intracavity injection of 131I monoclonal antibody: feasibility pharmacokinetics and dosimetry. Br J Cancer 1993;67:144–151.
74. Hopkins K, Chandler C, Bullimore J, et al. A pilot study of the treatment of patients with recurrent malignant gliomas with intratumoral yttrium-90 radioimmunoconjugates. Radiother Oncol 1995;34(2):121–131.
75. Thurber GM. Kinetics of antibody penetration into tumors. In: Speer TW, ed. Targeted radionuclide therapy. Philadelphia: Lippincott Williams & Wilkins, 2011:168–181.
76. Hopkins K, Chandler C, Eatough J, et al. Direct injection of 90Y MoAbs into glioma tumor resection cavities leads to limited diffusion of the radioimmunoconjugates into normal brain parenchyma: a model to estimate absorbed radiation dose. Int J Radiation Oncol Biol Phys 1998;40:835–844.
77. Mamelak AN, Rosenfeld S, Bucholz R, et al. Phase I single-dose study of intracavitary-administered iodine-131-TM-601 in adults with recurrent high-grade glioma. J Clin Oncol 2006;24:3644–3650.
78. Akabani G, Reardon DA, Coleman RE, et al. Dosimetry and radiographic analysis of 131I-labeled anti-tenascin 81C6 murine monoclonal antibody in newly diagnosed patients with malignant gliomas: a phase II study. J Nucl Med 2005;46:1042–1051.
79. Reardon DA, Akabani G, Coleman RE, et al. Phase II trial of murine 131I-labeled antitenascin monoclonal antibody 81C6 administered into surgically created resection cavities of patients with newly diagnosed malignant gliomas. J Clin Oncol 2002;20:1389–1397.
80. Reardon DA, Akabani G, Coleman RE, et al. Salvage radioimmunotherapy with murine iodine-131-labeled antitenascin monoclonal antibody 81C6 for patients with recurrent primary and metastatic malignant brain tumors: phase II study results. J Clin Oncol 2006;24:115–122.
81. Reardon DA, Zalutsky MR, Akabani G, et al. A pilot study: 131I-antitenascin monoclonal antibody 81C6 to deliver a 44-Gy resection cavity boost. Neuro Oncol 2008;10(2):182–189.
82. Zalutsky MR, Reardon DA, Akabani G, et al. Clinical experience with α-particle-emitting 211At: treatment of recurrent brain tumor patients with 211At-labeled chimeric antitenascin monoclonal antibody 81C6. J Nucl Med 2008;49(1):30–38.
83. McLendon RE, Akabani G, Friedman HS, et al. Tumor resection cavity administered iodine-131-labeled antitenascin 81C6 radioimmunotherapy in patients with malignant glioma: neuropathology aspects. Nucl Med Biol 2007;34:405–413.
84. Epenetos AA, Hird V, Lambert H, et al. Long term survival of patients with advanced ovarian cancer treated with intraperitoneal radioimmunotherapy. Int J Gynecol Cancer 2000;10:44–46.
85. Verheijen RH, Massuger LF, Benigno BB, et al. Phase III trial of intraperitoneal therapy with yttrium-90-labeled HMFG1 murine monoclonal antibody in patients with epithelial ovarian cancer after a surgically defined complete remission. J Clin Oncol 2006;24:571–578.
86. Oei AL, Verheijen RH, Seiden MV, et al. Decreased intraperitoneal disease recurrence in epithelial ovarian cancer patients receiving intraperitoneal consolidation treatment with yttrium-90-labeled murine HMFG1 without improvement in overall survival. Int J Cancer 2007;120:2710–2714.
87. Wallner PE. Unconjugated radiopharmaceuticals. In: Speer TW, ed. Targeted radionuclide therapy. Philadelphia: Lippincott Williams & Wilkins, 2011:294–297.
88. Wallner PE. “Naked” radiopharmaceuticals. Int J Radiation Oncology Biol Phys 2006;6(2 Suppl):S60–S61.
89. U.S. Food and Drug Administration. Radiation-emitting products. Available at: http://www.fda.gov/Radiation-EmittingProducts/default.htm. Accessed December 16, 2011.
90. U.S. NRC. Directory of agreement state and non-agreement state directors and state liaison officers. Available at: http://nrc-stp.ornl.gov/asdirectory.html. Accessed December 21, 2011.
91. U.S. NRC. Part 35—medical use of byproduct material. Available at: http://www.nrc.gov/reading-rm/doc-collections/cfr/part035/. Accessed December 16, 2011.
92. Perez-Monte JE. The U.S. NRC and the ADR process learning experiences. Health Phys 2011;101(Suppl 3):S160–S163.
93. Reisfield GM, Silberstein EB, Wilson GR. Radiopharmaceuticals for the palliation of painful bone metastases. Am J Hosp Palliat Care 2005;22:41–46.
94. Hindorf C, Flux GD, Ibisch C, et al. Clinical dosimetry in the treatment of bone tumors: old and new agents. Q J Nucl Med Mol Imaging 2011;55:198–204.
95. Jansen DR, Krijger GC, Kolar ZI, et al. Targeted radiotherapy of bone malignancies. Curr Drug Discov Technol 2010;7:233–246.
96. Liepe K. Alpharadin, a 223Ra-based alpha-particle-emitting pharmaceutical for the treatment of bone metastases in patients with cancer. Curr Opin Investig Drugs 2009;10:1346–1358.
97. Zafeirakis A, Zissimopoulos A, Baziotis N, et al. Introduction of a new semi-quantitative index with predictive implications in patients with painful osseous metastases after (186)Re-HEDP therapy. Q J Nucl Med Mol Imaging 2011;55:91–102.
98. Biersack HJ, Palmedo H, Andris A, et al. Palliation and survival after repeated (188)Re-HEDP therapy of hormone-refractory bone metastases of prostate cancer: a retrospective analysis. J Nucl Med 2011;52:1721–1726.
99. Liu C, Brasic JR, Liu X, et al. Timing and optimized acquisition parameters for the whole-body imaging of 177Lu-EDTMP toward performing bone pain palliation treatment. Nucl Med Commun 2012;33:90–96.
100. Ogawa K, Kawashima H, Shiba K, et al. Development of [(90)Y]DOTA-conjugated bisphosphonate for treatment of painful bone metastases. Nucl Med Biol 2009;36:129–135.
101. Das T, Chakraborty S, Sarma HD, et al. (170)Tm-EDTMP: a potential cost-effective alternative to (89)SrCl(2) for bone pain palliation. Nucl Med Biol 2009;36:561–568.
102. Silberstein EB. Teletherapy and radiopharmaceutical therapy of painful bone metastases. Sem Nuc Med 2005;35:152–158.
103. Nair N. Relative efficacy of 32P and 89Sr in palliation of skeletal metastases. J Nucl Med 1999;40:256–261.
104. Pecher P. Biological investigation with radioactive calcium and strontium: preliminary report on the use of radioactive strontium in the treatment of metastatic bone cancer. Univ Calif Publ Pharmacol 1942;11:117–149.
105. Roque I, Figuls M, Martinez-Zapata MJ, et al. Radioisotopes for metastatic bone pain. Cochrane Database Syst Rev 2011;(6):CD003347.
106. Dillehay GL, Ellerbroek NA, Balon H, et al. Practice guideline for the performance of therapy with unsealed radiopharmaceutical sources. Int J Radiat Oncol Biol Phys 2006;64:1299–1307.
107. Lutz S, Berk L, Chang E, et al. Palliative radiotherapy for bone metastases: an ASTRO evidence-based guideline. Int J Radiat Oncol Biol Phys 2011;79:965–976.
108. Sawin CT, Becker DV. Radioiodine and the treatment of hyperthyroidism: the early history. Thyroid 1997;7:163–176.
109. Mattsson S, Johansson L, Jonsson H, et al. Radioactive iodine in thyroid medicine—how it started in Sweden and some of today’s challenges. Acta Oncologica 2006;45:1031–1036.
110. International Atomic Energy Agency. Nuclear medicine in thyroid cancer management: a practical approach. Vienna, Austria: International Atomic Energy Agency, 2009.
111. Seidlin SM, Marrinelli LD, Oshry E. Radioactive iodine therapy: effect on functioning metastases and adenocarcinoma of thyroid. JAMA 1946;132:838–847.
112. Kulkarni K, Van Nostrand D, Atkins F. 131-I ablation and treatment of well-differentiated thyroid cancer. In: Speer TW, ed. Targeted radionuclide therapy. Philadelphia: Lippincott Williams & Wilkins, 2011:281–293.
113. Reiners C, Dietlein M, Luster. Radio-iodine therapy in differentiated thyroid cancer: indication and procedure. Best Pract Res Clin Endocrinol Metab 2008;22:989–1007.
114. Dohan O, De la Vieja A, Paroder V, et al. The sodium/iodide symporter (NIS): characterization, regulation, and medical significance. Endocr Rev 2002;24:48–77.
115. Wong KK, Dvorak RA, Marzola MC, et al. Molecular imaging in the management of thyroid cancer. Q J Nucl Med Imaging 2011;55:541–559.
116. Mazzaferri EL, Jhiang SM. Long-term impact of initial surgical and medical therapy on papillary and follicular thyroid cancer. Am J Med 1994;97:418–428.
117. Samaan NA, Schultz PN, Hickey RC, et al. The results of various modalities of treatment of well differentiated thyroid carcinoma: a retrospective review of 1599 patients. J Clin Endocrinol Metab 1992;75:714–720.
118. Wong JB, Kaplan MM, Meyer KB, et al. Ablative radioactive iodine therapy for apparently localized thyroid carcinoma. A decision analytic perspective. Endocrinol Metab Clin North Am 1990;19:741–760.
119. Hay ID, McConahey WM, Goellner JR. Managing patients with papillary thyroid carcinoma: insights gained from the Mayo Clinic’s experience of treating 2,512 consecutive patients during 1940 through 2000. Trans Am Clin Climatol Assoc 2002;113:241–260.
120. Mazzaferri EL. Treating differentiated thyroid carcinoma: where do we draw the line? Mayo Clin Proc 1991;66:105–111.
121. Van De Velde CJ, Hamming JF, Goslings BM, et al. Report of the consensus development conference on the management of differentiated thyroid cancer in the Netherlands. Eur J Cancer Clin Oncol 1988;24:287–292.
122. Mazzaferri EL, Kloos RT. Using recombinant human TSH in the management of well-differentiated thyroid cancer: current strategies and future directions. Thyroid 2000;10:767–778.
123. Dorn R, Kopp J, Vogt H, et al. Dosimetry-guided radioactive iodine treatment in patients with metastatic differentiated thyroid cancer: largest safe dose using a risk-adapted approach. J Nucl Med 2003;44:451–456.
124. Siegel HJ, Luck JV Jr, Siegel M, et al. Advances in radionuclide therapeutics in orthopaedics. J Am Acad Orthop Surg 2004;12:55–64.
125. Soroa VE, del Huerto Velazquez Espeche M, Giannone C, et al. Effects of radiosynovectomy with p-32 colloid therapy in hemophilia and rheumatoid arthritis. Cancer Biotherm Radiopharm 2005;20:344–348.
126. Potter ME, Partridge EE, Shingleton HM, et al. Intraperitoneal chromic phosphate in ovarian cancer: risks and benefits. Gynecol Oncol 1989;32:314–318.
127. Soper JT, Berchuk A, Dodge R, et al. Adjuvant therapy with intraperitoneal chromic phosphate (32P) in women with early ovarian carcinoma after comprehensive surgical staging. Obstet Gynecol 1992;79:993–997.
128. Spanos WJ, Day T, Jose B, et al. Use of P-32 in stage III epithelial carcinoma of the ovary. Gynecol Oncol 1994;54:35–39.
129. Young RC, Brody MF, Nieberg RK, et al. Adjuvant treatment for early ovarian cancer: a randomized phase III trial of intraperitoneal 32P or intravenous cyclophosphamide and cisplatin—a Gynecologic Oncology Group study. J Clin Oncol 2003;21:4350–4355.
130. Order SE, Siegel JA, Lustig RA, et al. A new method of delivering radioactive cytotoxic agents in solid cancers. Int J Radiat Oncol Biol Phys 1994;30:715–720.
131. Firusian N, Dempke W. An early phase II study of intratumoral P-32 chromic phosphate injection therapy for patients with refractory solid tumors and solitary metastases. Cancer 1999;85;980–987.
132. Porter AT, McEwan AJ, Powe JE, et al. Results of a randomized phase-III trial to evaluate the efficacy of strontium-89 adjuvant to local field external beam irradiation in the management of endocrine resistant metastatic prostate cancer. Int J Radiat Oncol Biol Phys 1993;25:805–813.
133. Tu SM, Lin SH. Current trials using bone-targeting agents in prostate cancer. Cancer J 2008;14:35–39.
134. Sartor O. Overview of samarium Sm 153 lexidronam in the treatment of painful metastatic disease of bone. Rev Urol 2004;6(Suppl 10);S3–S12.
135. Anderson PM, Wiseman GA, Erlandson L, et al. Gemcitabine radiosensitization after high-dose samarium for osteoblastic osteosarcoma. Clin Cancer Res 2005;11:6895–6900.
136. Nilsson S, Laren RH, Fossa SD, et al. First clinical experience with α-emitting radium-223 in the treatment of skeletal metastases. Clin Cancer Res 2005;11:4451–4450.
137. Nilsson S, Franzen L, Parker C, et al. Bone-targeted radium-223 in symptomatic, hormone-refractory prostate cancer: a randomised, multicentre, placebo-controlled phase II study. Lancet Oncol 2007;8:587–594.
138. O’Sullivan JM, McCready VR, Flux G, et al. High activity rhenium-186 HEDP with autologous peripheral blood stem cell rescue: a phase I study in progressive hormone refractory prostate cancer metastatic to bone. Br J Cancer 2002;86:1715–1720.
139. Liepe K, Runge R, Kotzerke J. The benefit of bone-seeking radiopharmaceuticals in the treatment of metastatic bone disease. J Cancer Res Clin Oncol 2005;131:60–66.
140. Li S, Liu J, Zhang H, et al. Rhenium-188 HEDP to treat painful bone metastases. Clin Nuc Med 2001;26:919–922.
141. Zhang H, Tian M, Li S, et al. Rhenium-188-HEDP therapy for the palliation of pain due to osseous metastases in lung cancer patients. Cancer Biother Radiopharm 2003;18:719–726.
< div class='tao-gold-member'>




