Drug
Primary mode of action
Metabolism and platelet inhibition
Platelet inhibition
Dosing
Aspirin
COX-1 inhibition
Prodrug
Irreversible
Once daily
Clopidogrel
P2Y12 receptor antagonism
Prodrug
Irreversible
Once daily
Prasugrel
P2Y12 receptor antagonism
Prodrug
Irreversible
Once daily
Ticagrelor
Allosteric P2Y12 receptor antagonism
Direct-acting
Reversible
Twice daily
3.1 Pharmacology
Clopidogrel is a second-generation thienopyridine, which became available in its generic form in 2012. Clopidogrel is a prodrug, which is well absorbed from the gut, but remains pharmacologically inert until activated in the liver through the CYP system (Fig. 1). The majority of administered clopidogrel is metabolized by an esterase pathway not resulting in active drug metabolites, and only 15 % reaches the liver for active metabolite transformation [14]. This is mediated by a two-step oxidative process regulated by the CYP system. Ultimately, as little as 2 % ends up irreversibly inhibiting the P2Y12 receptor [21]. Among the different CYP variants involved in the hepatic conversion of clopidogrel, CYP2C19 is the major variant responsible for approximately 45 % [21].
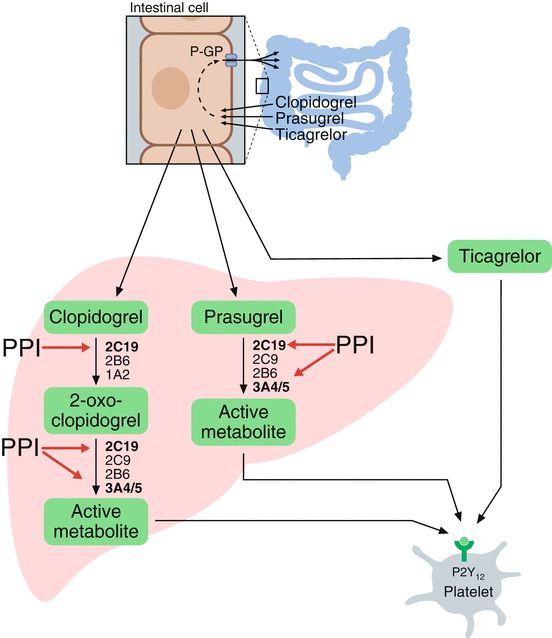
Fig. 1
A schematic presentation of the absorption and metabolism of clopidogrel, prasugrel, and ticagrelor (Adapted from Würtz et al. [112]). Clopidogrel is activated by a two-step oxidative process in the liver, whereas only one oxidative step is needed for the activation of prasugrel. The most important CYP enzymes mediating hepatic bioactivation of clopidogrel and prasugrel are depicted. CYP2C19 and CYP3A4/A5 are highlighted because they are strongly involved in the metabolism of certain PPIs, in particular omeprazole, thereby competitively inhibiting the bioactivation of clopidogrel and prasugrel. Ticagrelor does not require hepatic bioactivation. CYP cytochrome P450, P-GP P-glycoprotein (multidrug resistance protein), PPI proton pump inhibitor
Prasugrel is activated in a one-step oxidative process and, unlike clopidogrel, none of the drug is shunted to an inactive pathway (Fig. 1). Compared to clopidogrel, the hepatic conversion of prasugel is less dependent on CYP2C19 [22]. Ticagrelor is an adenosine triphosphate analogue not belonging to the thienopyridine family. Ticagrelor inhibits the P2Y12 receptor reversibly and does not require hepatic bioactivation (Table 1 and Fig. 1). Prasugrel and ticagrelor are more potent platelet function inhibitors than clopidogrel and are now being widely used in combination with aspirin in the setting of ACS.
3.2 Clinical Use
The CURE trial from 2001 documented the benefit of clopidogrel in addition to aspirin in patients with non-ST elevation MI [23]. The relative risk for the primary end point (cardiovascular death, non-fatal MI, or stroke) with aspirin and clopidogrel was 0.80 (95 % confidence interval [CI] 0.72–0.90) compared to aspirin alone. Since then, clopidogrel has been used in combination with aspirin in the setting of percutaneous coronary intervention (PCI), especially in the treatment of ACS. In 2005, a similar benefit was documented in patients with ST elevation MI [24, 25]. Overall, dual antiplatelet therapy with aspirin and clopidogrel in patients with ACS reduced cardiovascular risk by approximately 10 % compared to aspirin alone [23–25]. Documenting its widespread use, clopidogrel was the second most prescribed drug worldwide in 2010 (atorvastatin was the most prescribed) [26].
From 2009 to 2011 ticagrelor and prasugrel received authorization from European and American authorities for use in combination with aspirin for prevention of atherothrombotic events in patients with ACS undergoing PCI. Approvals were based on two phase III trials, TRITON-TIMI 38 (prasugrel) [27] and PLATO (ticagrelor) [28], documenting significant reductions in cardiovascular death, non-fatal MI, or stroke when using prasugrel or ticagrelor instead of clopidogrel. In TRITON-TIMI 38 the hazard ratio with prasugrel was 0.81 (95 % CI 0.73–0.90), and in PLATO the hazard ratio with ticagrelor was 0.84 (95 % CI 0.77–0.92). Although prasugrel and ticagrelor increased the risk of non-coronary artery bypass grafting-related major bleeding according to the Thrombolysis in Myocardial Infarction criteria (by 32 % and 25 %, respectively), both drugs are now widely used as treatment and short-term prevention of atherothrombotic events in patients with ACS [4].
4 Antiplatelet Treatment and Gastrointestinal Bleeding
Cardiovascular protection by aspirin and ADP receptor antagonists accrue at the expense of an increased risk of upper gastrointestinal bleeding [29, 30]. Gastrointestinal bleeding is life-threatening, especially in patients presenting with ACS [31] and documenting this, aspirin remains the dominant contributor to gastrointestinal bleeding-related mortality [32].
The gastrotoxic effects of aspirin that cause ulceration and bleeding have been attributed to (1) topical mucosal injury caused by inhibition of prostaglandin and (2) systemic antiplatelet effects driven by inhibition of thromboxane A2 generation [33, 34]. Prostaglandins are essential in protecting the gastric mucosa. They increase mucosal blood flow, promote proliferation of gastric epithelial cells, and stimulate mucus and bicarbonate secretion. Therefore, inhibition of prostaglandin synthesis by aspirin makes the gastric mucosa susceptible to ulcer formation and bleeding in the highly acidic environment. Furthermore, platelet inhibition with aspirin impairs healing of the vulnerable gastric mucosa [33, 34].
5 Proton Pump Inhibitors: Pharmacology and Clinical Use
Strategies to prevent gastrointestinal discomfort, ulceration, and bleeding during antiplatelet treatment include the identification and modification of associated risk factors as well as concomitant treatment with gastroprotective agents, mainly histamine H2 receptor antagonists and PPIs [33, 35]. For more than two decades, PPIs have been used extensively for the treatment of gastric acid-related disorders. Even though H2 receptor antagonist are effective in preventing gastrointestinal complications [36], PPIs produce a higher degree and longer duration of gastric acid suppression than H2 receptor antagonists leading to higher healing rates [8]. Although PPIs have widely been considered harmless, there are studies associating these drugs with serious adverse effects such as pneumonia, interstitial nephritis, osteoporotic fractures, and intestinal Clostridium difficile infections [37].
Under acidic conditions, PPIs are protonated and converted to cyclic sulphenamides. These active PPI metabolites reduce gastric acid production by irreversibly inhibiting the enzyme responsible for gastric acid secretion: the H+/K+-exchanging adenosine triphosphatase, often referred to as “the proton pump” [8]. The proton pump, which is located on gastric parietal cells, is directly responsible for H+ secretion into the gastric lumen. It follows that PPIs, as opposed to H2 receptor antagonists, target the terminal step in gastric acid secretion making the gastric acid suppression particularly strong. PPIs have a short plasma half-life of 30–120 min depending on pH level, yet the antacid effect is sustained for days due to the irreversible inhibition as well as accumulation of the drug in parietal cells [8].
6 Biochemical Background for Putative Drug Interactions Between Proton Pump Inhibitors and Antiplatelet Drugs
Under physiological conditions, aspirin is absorbed in its non-ionized lipid state across the gastric mucosal barrier. A pH-dependent mechanism has been suggested to explain a drug interaction between aspirin and PPIs. PPI reduce gastric acid production by inhibiting the enzyme responsible for gastric acid secretion from gastric parietal cells: the H+/K+-exchanging adenosine triphosphatase (Fig. 2) [103]. According to the pH partition hypothesis [38], modifying the intragastric milieu by raising pH potentially reduces the bioavailability of drugs, in particular those being absorbed across the gastric mucosal membrane, such as aspirin [39]. During PPI treatment, intragastric pH does indeed rise above the pK a (3.5) of aspirin potentially reducing its lipophilicity and gastric absorption [39, 40].
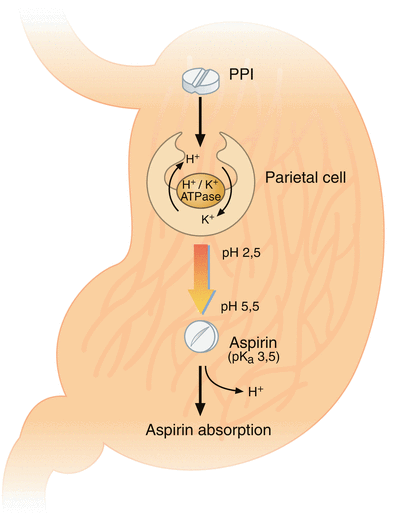
Fig. 2
Suggested biochemical background for a drug interaction between aspirin and proton pump inhibitors (Adapted from Würtz and Grove [113]). Under normal physiological conditions, aspirin is absorbed in its non-ionized lipid state across the gastric mucosal barrier. Proton pump inhibitors inhibit the H+/K+-exchanging ATPase of the gastric parietal cells. Intragastric pH rises above the pK a (3.5) of aspirin and reduces the lipophilicity of aspirin thereby lowering its gastric absorption. ATP adenosine triphosphate, PPI proton pump inhibitor
The activity of CYP2C19 is altered by PPIs, which are CYP2C19 substrates and thus may interact with clopidogrel and prasugrel metabolism through competitive antagonism. It follows that the interaction between PPIs and thienopyridines depends on the capacity of each PPI subtype to inhibit CYP2C19. Omeprazole, esomeprazole, and lansoprazole have a relatively high potency towards CYP2C19, while rabeprazole and pantoprazole have less potency. Accordingly, PPIs with low inhibitory effect on CYP219 are recommended if combined treatment with a thienopyridine and a PPI is required [35].
7 Interactions Between Proton Pump Inhibitors and Aspirin
The number of studies addressing a drug interaction between PPIs and aspirin remains relatively sparse (Table 2). Evidence is gathered from statistical modeling [41], pharmacokinetic measurements [42–44], large observational studies with clinical end points [45, 46], post–hoc analyses of large clinical trials [47], smaller interventional studies with clinical end points [48], or derived from studies utilizing ex vivo platelet function tests as a marker for the clinical effect of aspirin [49–53].
Table 2
Studies evaluating the association between proton pump inhibitor use and the antiplatelet effect of aspirin
Study | Participants (n) | Antiplatelet treatment | PPI type (n) | Design | Test/end point | Main results |
|---|---|---|---|---|---|---|
Iñarrea et al. [49] | Healthy volunteers (14) | Non enteric-coated ASA 125 mg ± PPI | Omeprazole | Cross-over study | Skin bleeding time, light transmittance aggregometry (Aggrecorder), plasma levels of ASA and salicylic acid | Omeprazole did not affect bleeding time, platelet aggregation, or plasma levels of ASA and salicylic acid |
Adamopoulos et al. [51] | Hypertensive subjects with indication for primary prophylaxis with aspirin | Enteric-coated ASA 100 mg ± PPI | Lansoprazole | Cross-over study | Light transmittance aggregometry (APACT4) and closure time (PFA-100) | Platelet aggregation and closure time during ASA treatment were unaffected by lansoprazole |
Niazi et al. [42] | Healthy volunteers (55) | Non enteric-coated ASA 325 mg ± PPI | Esomeprazole | Cross-over study | Maximum ASA plasma concentration and steady-state area under the concentration-time curve | Pharmacokinetic measures of ASA were unaffected by esomeprazole |
Kasprzak et al. [50] | PCI for ACS (31) | Enteric-coated ASA 75 mg ± PPI | Pantoprazole | Cross-over study | Platelet aggregometry (Multiplate Analyzer) | Reduced platelet aggregation in PPI users compared to non-users |
Andersson et al. [53] | Healthy volunteers (29) | ASA 81 mg (coating not specified) ± PPI | Esomeprazole | Cross-over study | Optical aggregometry (VerifyNow Aspirin) and thromboxane B2 | The drop in platelet aggregation and thromboxane B2 from baseline (no aspirin) to post-treatment was equal whether treatment was ASA + PPI or ASA alone |
Würtz et al. [52] | Coronary artery disease (418) | Non enteric-coated ASA 75 mg | Pantoprazole, esomeprazole, omeprazole, and lansoprazole | Cohort study | Platelet aggregometry (Multiplate Analyzer) and platelet activation level (soluble P-selectin) | Increased platelet aggregation, platelet activation, and thromboxane B2 levels in PPI users compared to non-users |
PPI: 54 users, 364 non-users | ||||||
Charlot et al. [45] | 30-day survivors of a first-ever MI between 1997 and 2006 (19,925) | ASA (dose and coating not specified), clopidogrel users excluded | Pantoprazole, omeprazole, lansoprazole, and esomeprazole | Retrospective nationwide register-based study | 1-year composite of cardiovascular death, MI, or stroke | PPI use, but not H2 receptor antagonist use, at any time following discharge associated with increased risk of the composite end point compared with non-use in the multivariable adjusted (multivariable adjusted hazard ratio 1.46, 95 % CI 1.33–1.61; propensity score-matched hazard ratio 1.61, 95 % CI 1.45–1.79) |
Dunn et al. [47] | Post-hoc analysis of the CAPRIE trial | ASA 325 mg or CLO 75 mg | Omeprazole and lansoprazole | Post-hoc analysis of a clinical trial | 1-year composite of ischemic stroke, MI, or vascular death | In ASA-treated patients, PPI use was not associated with an increased risk of the composite end point compared to non-use (adjusted hazard ratio 1.04, 95 % CI 0.70–1.57) |
Previous MI, ischemic stroke, or peripheral vascular disease (ASA arm, 9586) | ||||||
Whellan et al. [48] | Users of ASA for secondary cardiovascular prevention (1049) | Enteric-coated ASA 325 mg ± PPI | Omeprazole | Randomized PPI assignment | 6-month major adverse cardiovascular events and upper gastrointestinal symptoms | The rate of cardiovascular events was equal between treatment arms, but upper gastrointestinal symptoms were reduced with the combination tablet compared to ASA alone |
PPI was given as a single-tablet combination of ASA and omeprazole | ||||||
Garcia Rodriguez et al. [46] | Users of ASA for secondary cardiovascular prevention (39,513) or in patients with previous ACS (42,542) between 2000 and 2007 | ASA 75–300 mg (>85 % received 75 mg, coating not specified) | Not specified | Cohort study | Composite of non-fatal MI or coronary death | PPI use not associated with increased risk of non-fatal MI or coronary death in neither of the study cohorts (pooled relative risk 0.96, 95 % CI 0.62−1.48) |
In previous animal studies, omeprazole reduced the analgesic and antipyretic effects of aspirin, which was measured by means of reduced gastric aspirin absorption [40, 54]. Similar findings were reported from a study of humans [55]. On the other hand, Iñarrea et al. measured the antiplatelet effect of aspirin in 14 healthy individuals before and after 4 days of 20 mg/day omeprazole treatment. Bleeding time and platelet aggregation levels were both unaffected by omeprazole [49]. In a randomized cross-over study of 24 healthy individuals, 100 mg of enteric-coated aspirin was given for 4 weeks with or without concomitant 30 mg/day lansoprazole. Thereafter, participants were switched to the other treatment regimen for another 4 weeks. Platelet function assessed by light transmittance aggregometry (APACT 4) and shear stress-stimulated closure time (Platelet Function Analyzer-100) suggested no difference in antiplatelet potency between aspirin with lansoprazole and aspirin alone [51]. Another study showed no pharmacokinetic interaction based on measurements of acetylsalicylic acid plasma concentrations in 55 healthy volunteers subjected to three treatment periods comprising esomeprazole, aspirin, and both [42]. Subsequently, the authors evaluated the bioequivalence between 40 mg esomeprazole and 325 mg aspirin given separately and as a single-tablet formulation including both agents. Analyzing the same end point of acetylsalicylic acid maximal plasma concentration, the two treatment schemes remained bioequivalent [43]. In a randomized cross-over study, 29 healthy individuals received low-dose aspirin with or without esomeprazole 20 mg once daily for 5 days followed by 14-day washout and subsequent treatment cross-over. Platelet aggregation evaluated with the VerifyNow® Aspirin test did not differ between the two treatment regimens, neither did levels of serum thromboxane B2 [53].
In a pharmacodynamic study by Würtz et al., we included 418 aspirin-treated patients with stable coronary artery disease, of whom 54 were PPI users. In multivariable adjusted analyses, platelet aggregation (median 180 [interquartile range 119–312] vs. 152 [84–226] aggregation units*minute, p = 0.013) and platelet activation measured by soluble serum P-selectin (88.5 [65.2–105.8] vs 75.4 [60.0–91.5] ng/ml, p = 0.013) were significantly higher in patients treated with a PPI. In contrast to many other pharmacodynamic studies, a non-enteric coated formulation of aspirin was used in this study, which may be important given that gastric absorption of enteric-coated aspirin has been shown to increase during omeprazole-treatment [56]. The findings by Würtz et al. were supported by a large Danish register-based study of 19,925 patients suffering a first-time MI. All patients were treated with aspirin, while almost 30,000 patients treated with clopidogrel were excluded. The risk of cardiovascular death, recurrent MI, or stroke was increased in patients receiving a PPI (adjusted hazard ratio 1.46, 95 % CI 1.33–1.61), but not in patients receiving a gastroprotective H2 receptor antagonist [45].
Whellan et al. tested the hypothesis that a single-tablet formulation (PA32540) [57] of enteric-coated aspirin (325 mg) and immediate-release omeprazole (40 mg) would reduce gastrointestinal complications without promoting thrombotic complications compared to aspirin alone. A coordinated-delivery tablet was used, in which omeprazole is embedded within a film coat enabling instantaneous dissolution, whereas aspirin release occurs only when gastrointestinal pH reaches a level of 5.5 [48]. The primary end point of endoscopically verified gastric ulcer at 6 months occurred less frequently among users of the combined formulation (3.2 % vs. 8.6 %, p < 0.001), while the rate of major adverse cardiovascular events did not differ between treatment arms (1.7 % vs. 2.5 %, p > 0.05). Importantly, the study had a low rate of cardiovascular events, for which the study was underpowered [48].
Most recently, the combined analysis of coronary event rates in two large cohorts of first-time users of aspirin for secondary prevention was published [46]. The first cohort included first-time users of aspirin for any secondary prevention indication, while the second cohort consisted of patients who initiated aspirin treatment following an acute coronary event. Looking at the cohorts separately or combined, PPI treatment was not associated with an increase in the risk of non-fatal MI or coronary death [46], and the results thus contrast those of the above mentioned large registry-based study [45].
A recent analysis showed that co-prescription of low-dose aspirin and a PPI turned out to be cost-effective by reducing gastrointestinal as well as cardiovascular events [41]. This cost-effectiveness analysis was based on previously published clinical studies, and the cardiovascular benefit appeared to be partly driven by increased adherence to aspirin in PPI users. Furthermore, even in patients with cardiovascular disease who continue aspirin treatment after suffering a gastrointestinal bleeding event, aspirin seems to confer a net clinical benefit because the risk of bleeding is outbalanced by improved cardiovascular outcome [58]. This was shown in a small randomized study, in which aspirin users who suffered a peptic ulcer bleeding were given either aspirin or placebo on top of pantoprazole. While increasing the risk for recurrent gastrointestinal bleeding, continued aspirin treatment reduced mortality [58]. Although these interesting results should be confirmed in larger studies, they stress that discontinuing aspirin upon gastrointestinal events should be carefully considered in patients with increased risk of cardiovascular events.
Altogether, studies exploring whether PPIs reduce the effect of aspirin are sparse. Studies are small and relatively heterogeneous and this, coupled with the fact that only one randomized, yet underpowered, study has been performed makes it premature to change clinical recommendations at present as reflected in current guidelines [4, 5, 35].
8 Interaction Between Proton Pump Inhibitors and Clopidogrel
8.1 Pharmacological Studies
Since 2006, several observational studies have reported an attenuation of the antiplatelet effect of clopidogrel when given concomitantly with PPI, particularly omeprazole (Table 3). Gilard et al. used the vasodilator-stimulated phosphoprotein (VASP) phosphorylation assay to assess platelet function 48 h after treatment initiation in 105 patients undergoing angiography. All patients were treated with aspirin and clopidogrel, and 24 patients were also treated with a PPI. PPI users had a significantly higher platelet reactivity index than non-users (61.4 ± 23.2 % vs. 49.5 ± 16.3 %, p = 0.007) [59]. Indeed, the VASP assay reflects the extent of intracellular P2Y12 pathway inhibition and is therefore considered the pharmacologically most specific test of platelet inhibition by ADP receptor antagonists [60]. Pursuing more firm documentation, the authors conducted the double-blind placebo-controlled OCLA trial published in 2008 [61]. A total of 124 patients undergoing PCI received standard doses of aspirin and clopidogrel and were randomized to either omeprazole 20 mg/day or placebo for 7 days. Platelet inhibition was assessed at days one and seven using the platelet reactivity VASP index. On day seven, the omeprazole-arm had significantly higher platelet reactivity than the placebo-arm (51.4 ± 16.4 % vs. 39.8 ± 15.4 %, p < 0.0001) [61]. Given the rigorous design of the OCLA trial, the results were convincing, and many, but not all [62], subsequent studies supported the findings [63–69].
Table 3
Studies with pharmacokinetic and/or pharmacodynamic end points suggesting an association between proton pump inhibitor use and the antiplatelet effect of clopidogrel
Study | Participants (n) | Antiplatelet treatment | PPI type (n) | Random PPI assignment? | Test | Main results |
|---|---|---|---|---|---|---|
Gilard et al. [59] | High-risk coronary angioplasty (105) | ASA and CLO for a minimum of 48 h | Not specified | No | VASP-PRI | Increased PRI in PPI users (61.4 ± 23.2 vs. 49.5 ± 16.3, p = 0.007) |
PPI: 24 users, 81 non-users | No association found with statins, angiotensin converting enzyme inhibitors, angiotensin II receptor antagonist, or beta blockers | |||||
Gilard et al. [61] | Elective PCI (124) | ASA and CLO + PPI or placebo (7 days) | Omeprazole | Yes | VASP-PRI | Increased PRI in the PPI-arm at day 7 (39.8 ± 15.4 % vs. 51.4 ± 16.4 %), but not at day 1 (83.2 ± 5.6 % vs. 83.9 ± 4.6 %, not significant) |
Small et al. [63] | Healthy volunteers (24) | CLO 300 mg or PRA 60 mg ± PPI | Lansoprazole | Yes (cross-over design) | Pharmacokinetics: Maximum CLO plasma concentration and area under the concentration-time curve | PPI use did not affect the pharmacokinetics of CLO, but tended to reduce maximal inhibition of platelet aggregation, which was most pronounced in subjects with aggregation levels |
Pharmacodynamics: Optical platelet aggregometry | ||||||
O’Donoghue et al. [64] | Elective PCI (201). Post-hoc analysis of PRINCIPLE-TIMI 44 | Random assignment to CLO 600 mg LD/300 mg MD or PRA 60 mg LD/10 mg MD | Not specified | No | Light transmittance aggregometry | Reduced platelet inhibition in CLO-treated PPI users compared to non-users 2, 6, and 18–24 h after PCI |
2 h: 10.4 ± 16.2 % vs. 24.2 ± 20.5 %, p = 0.003 | ||||||
6 h: 23.2 ± 19.5 % vs. 35.2 ± 20.9 %, p = 0.02 | ||||||
18–24 h: 23.8 ± 14.4 % vs. 36.1 ± 20.8 %, p = 0.03 | ||||||
No differences 30 min nor 15 days after PCI | ||||||
Sibbing et al. [65] | Scheduled for control angiography after PCI (1000) | ASA 75 mg and CLO 75 mg | Pantoprazole, omeprazole, and esomeprazole | No | Platelet aggregometry (Multiplate Analyzer) | Increased platelet aggregation in omeprazole users compared to non-users (295.5 [IQR 193.5–571.2] aggregation units*min vs. 220.0 [IQR 143.8–388.8] aggregation units*min; p = 0.001). No differences between pantoprazole/esomeprazole users and PPI non-users |
Zuern et al. [67] | Elective or urgent PCI (1425) | ASA 100 mg and CLO 600 mg LD/75 mg MD | Pantoprazole, esomeprazole, and omeprazole | No | Optical aggregometry (Chronolog Lumi Aggregometer) | Increased platelet aggregation in PPI users compared to non-users (34.0 ± 21.4 % vs. 29.8 ± 20.2 %, p < 0.001). PPI use was an independent predictor of residual platelet aggregation |
Cuisset et al. [66] | PCI for non-ST elevation MI (104) | ASA 250 mg LD/75 mg MD and CLO 600 mg LD/150 mg MD plus PPI | Omeprazole and pantoprazole | Yes | VASP-PRI and optical aggregometry (PAP4 Aggregometer) | 12–24 h after LD: No differences in terms of PRI or aggregation |
1 month after discharge: Increased PRI in omeprazole users compared to pantoprazole users (36 ± 20 % vs. 48 ± 17 %, p = 0.007). Using PRI, more CLO non-responders among omeprazole users than pantoprazole users (44 % vs. 23 %, p = 0.04. Odds ratio 2.6, 95 % CI 1.2–6.2). No differences found by aggregometry | ||||||
Fontes-Carvalho et al. [68] | PCI for MI (34) | ASA 150 mg and CLO 75 mg plus PPI | Omeprazole or pantoprazole (cross-over, 1 month washout) | Yes (cross-over design) | Optical aggregometry (VerifyNow P2Y12) | 1 month after PCI: Significant increase in platelet aggregation during omeprazole-treatment compared to PPI non-use (235 ± 58 PRU vs. 201 ± 48, p < 0.001). The number of clopidogrel non-responders almost doubled during omeprazole-treatment. No differences seen with pantoprazole compared to PPI non-use |
Angiolillo et al. [69] | Healthy volunteers (282) | CLO 300 mg LD/75 mg MD | Omeprazole or pantoprazole | Yes (cross-over design) | VASP-PRI and platelet aggregometry | Significant increase in platelet aggregation and PRI during omeprazole-treatment compared to PPI non-use. The drug interaction was not mitigated by increasing clopidogrel dose or administering clopidogrel and omeprazole apart (spaced administration). Pantoprazole only affected platelet aggregation and VASP-PRI sparsely compared to omeprazole |
Parri et al. [73] | Primary PCI for ST elevation MI (105) | ASA 100 mg and CLO 300 mg LD/75 mg MD and optional glycoprotein IIb/IIIa inhibition | Pantoprazole or ranitidine (H2 receptor antagonist) | Yes | Light transmittance aggregometry and closure time (PFA-100) | Increased maximal platelet aggregation stimulated with ADP in pantoprazole users compared to ranitidine users after correction for CYP2C19*2 genotype both five (median 29 % vs. 19 %, p = 0.01) and 30 days (median 35 % vs. 27 %, p = 0.03) after PCI. No difference observed with other agonists or with the PFA-100 |
Of interest, some studies suggested a differential impact of proton pump inhibitors on the antiplatelet effect of clopidogrel. Four studies independently argued in favor of preferentially using non-omeprazole PPIs, namely pantoprazole, to avoid a drug interaction [65, 66, 68, 69]. In the PACA study, a total of 104 patients with non-ST elevation ACS were randomized to omeprazole or pantoprazole on top of aspirin and clopidogrel. After 1 month, platelet inhibition assessed by the VASP index was significantly greater with clopidogrel in patients receiving pantoprazole (36 ± 20 % vs. 48 ± 17 %, p < 0.007) [66].
Angiolillo et al. performed a complex study including four randomized, placebo-controlled, cross-over studies among 282 healthy individuals. The purpose was (1) to explore any drug interaction between clopidogrel and omeprazole, (2) to test if such interaction could be mitigated by administering clopidogrel and omeprazole 12 h apart, (3) or by doubling the clopidogrel maintenance dose to 150 mg daily, and (4) to compare the drug interaction caused by omeprazole with that caused by pantoprazole. Essentially, the study showed that omeprazole, but not pantoprazole, reduced the pharmacodynamic effect of clopidogrel through a pH-independent mechanism mediated by the CYP2C19 enzyme [69]. Since all PPIs lower gastric pH to roughly the same extent at equipotent doses [70, 71], the differential impact of PPIs on the platelet inhibitory effect of clopidogrel may rather be attributable to differences in the inhibitory potency towards CYP2C19. In particular, pantoprazole seems to interfere little, if at all, with the metabolism of clopidogrel and is known to have very little affinity for CYP2C19 [72]. Notwithstanding, a recent study suggested that pantoprazole increases platelet aggregation irrespective of CYP2C19*2 genotype in clopidogrel-treated patients with ST elevation MI undergoing PCI [73]. According to a post-hoc subgroup analysis of the PRINCIPLE-TIMI 44 trial, treatment with a PPI and clopidogrel increased the number of non-responders to a clopidogrel loading dose in the acute phase and to a 150 mg daily maintenance dose 15 days after PCI [64].
Related posts:
 Clinical Presentation and Basic Work up in Patients with Suspected Pulmonary Embolism
Prevention of Venous Thromboembolism in Surgical Patients
Clinical Presentation and Basic Work up in Patients with Suspected Pulmonary Embolism
Prevention of Venous Thromboembolism in Surgical Patients
 Diagnosis and Management of Early Deep Vein Thrombosis
Diagnosis and Management of Early Deep Vein Thrombosis
 Thrombosis: Regulatory Mechanisms and Emerging Directions
Thrombosis: Regulatory Mechanisms and Emerging Directions
 Venous Thrombosis
Venous Thrombosis
 Treatment of Thrombosis and Embolism
Treatment of Thrombosis and Embolism
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


