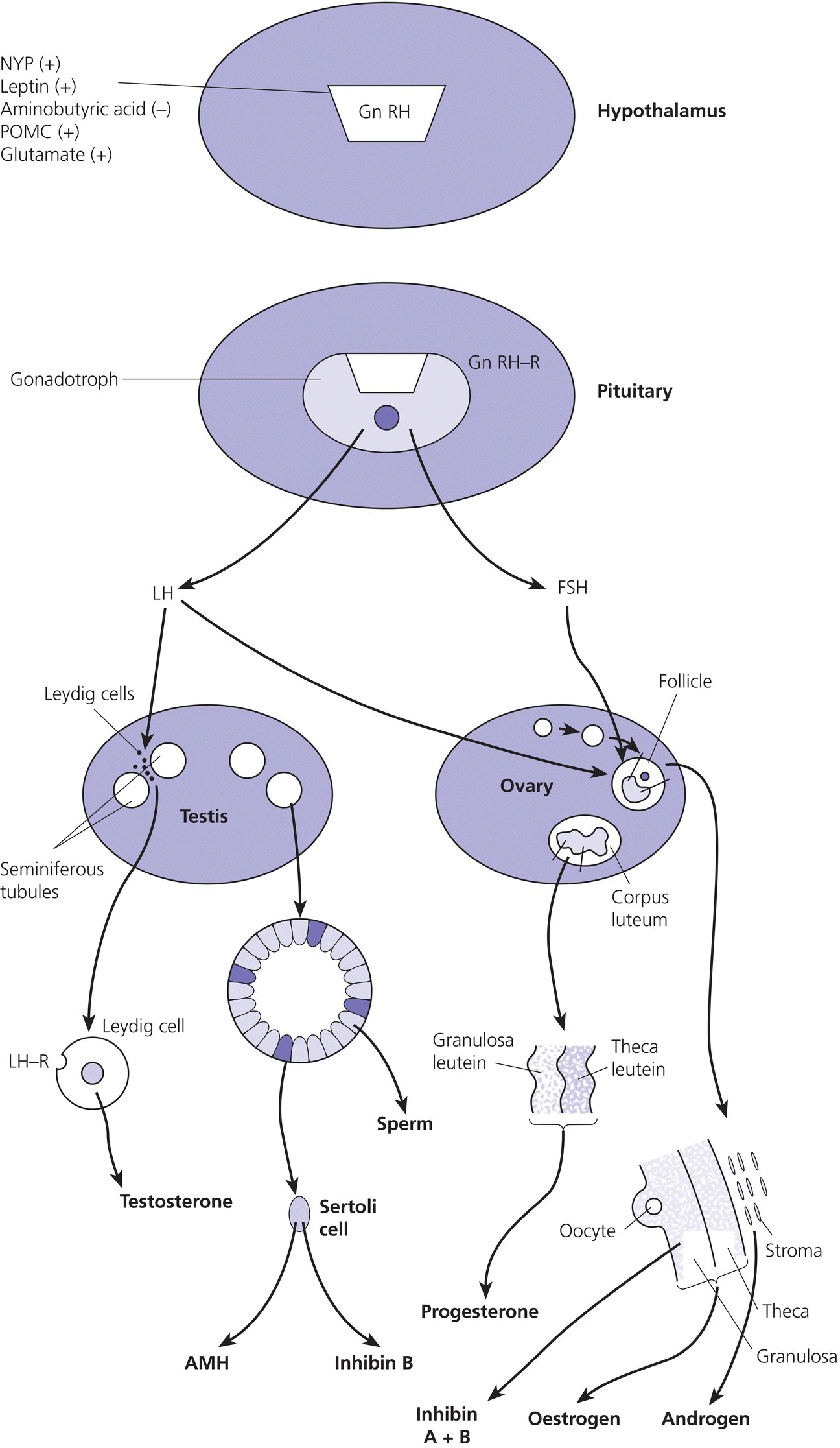
5 Puberty occurs when the secretion of gonadotrophin‐releasing hormone (GnRH) by the hypothalamus, which is largely but not entirely suppressed during childhood, increases so that pulsatile secretion of luteinizing hormone (LH) increases, resulting in sufficient sex steroid production to induce secondary sexual development (see Figure 5.1). Figure 5.1 Schematic representation of hypothalamic–pituitary–gonadal pathways. (See also Figure 3.4 of Chapter 3). Abbreviations: +, stimulatory effect; −, inhibitory effect; NPY, neuropeptide Y; POMC, pro‐opiomelanocortin C; GnRH, gonadotrophin‐releasing hormone; LH, luteinizing hormone; FSH, follicle‐stimulating hormone; LH‐R, luteinizing hormone receptor; AMH, anti‐Müllerian Hormone. During embryogenesis the primordial germ cells migrate to the ovary and develop into primordial follicles. In the foetus, the pool of primordial follicles reaches a peak of around 7 million germ cells at 20 weeks gestation, falling to 1–2 million at birth, 500000 at menarche, and about 100 at menopause. FSH levels reflect the size of the primordial follicle pool at any given time. Each follicle contains an oocyte arrested in the prophase of the first meiotic division. Follicular Phase (Approximately 14 Days, but Variable in Duration) Luteal Phase (Always 14 Days) The GnRH‐secreting neurons are under the influence of controlling neurons of both an excitatory (glutamate) and an inhibitory (gamma‐aminobutyric acid, GABA) nature. In 1999, a paternally imprinted gene was described, encoding Makorin ring finger protein 3 (MKRN3) (Jong et al. 1999). This protein has an inhibitory effect on GnRH secretion which wanes with the onset of puberty. GnRH neurons are also under the control of glial cells which are excitatory in effect. At the time of puberty, changes in trans‐synaptic neurotransmission and glial inputs (such as that provided by prostaglandins released by astrocytes) result in the pulsatile secretion of GnRH and hence the pubertal cascade. Two points should be noted: This involves an assessment of breast (B) development in girls, genital (G) development and testicular volume in boys, and pubic (P) and axillary (A) hair development in both sexes. This is the staging system, modified after Tanner and with Tanner’s original pictures shown in Figure 5.2. Figure 5.2 Tanner staging showing breast stages 1–5, pubic hair and genital stages 2–5. See text for key to stages (Tanner, 1962). N.B. Testicular volume should be gauged using the Prader orchidometer. The orchidometer allows adolescents who refuse to be examined to self‐assess their testicular volume. If an orchidometer is not available, length and breadth can be measured using a paper tape measure and assuming that breadth and depth are the same and employing the formula for a prolate ellipsoid, volume can be calculated as ‘length × breadth × depth × π/6. Penile size (useful for serial staging and important if hypogonadism is suspected) is measured from the base of the penis to tip of the glans (not the foreskin). In obese individuals, it is important to push back the suprapubic fat to obtain the true penile length. Boys enter puberty roughly six months later than girls. Mean ages of breast (B) and genital (G) stages in girls and boys with corresponding height velocities including peak height velocity (PHV) are shown in Table 5.1 (girls) and Table 5.2 (boys). Table 5.1 Mean ages of breast (B) stages in girls with corresponding height velocities including peak height velocity (PHV). Source: Data from Tanner and Whitehouse (1976). Breast budding is accompanied by acceleration in growth rate with PHV at B2–3 and usually marked deceleration in height velocity after menarche. Table 5.2 Mean ages of genital (B) stages in boys with corresponding height velocities including peak height velocity (PHV). Source: Data from Tanner and Whitehouse (1976). At G2 growth rate is either the same as or slower than at G1. Increase in growth rate coincides with testicular volumes of 6–10 ml. In girls, breast budding is accompanied by acceleration in growth rate with PHV at B2–3 and usually marked deceleration in height velocity after menarche. In boys, at G2, growth rate is either the same as or slower than at G1. The increase in growth rate coincides with testicular volumes of 8–12 ml. In both sexes, the age of onset and duration of pubertal development are subject to marked individual variation. Early puberty is associated with a higher PHV, and delayed puberty with a lower PHV. The 13 cm difference in height between adult males and females relates to the following three factors: As for short stature examination, this involves meticulous auxology followed by the taking of a focused history and carrying out an appropriate physical examination which includes pubertal staging In the context of current concerns about child and adolescent safe‐guarding, it is very important that the clinician approaches the issue of pubertal staging in a sensitive, open and honest manner. After the history taking and before the clinician rises from his/her seat to conduct the examination, the family are informed of the desirability of examining underarm and pubic hair in both girls and boys; breast development in girls; and penis and testicles in boys. The clinician explains to the family why the examination is needed, indicates that this is a matter of good clinical practice, but stresses that it can only be done if the patient and family/carer agree. Where possible, girls should be examined by a trained female member of staff and boys by a trained male staff member (neither of whom need to be medical). Where this is not feasible, a male examiner offers a girl the choice of her mother, or a female nurse as chaperone, while a female examiner offers the choice of a male chaperone or the father. In practice, boys tend to prefer to be examined in privacy (i.e. behind the screen or curtain) but with the parent present in the room. If the patient is initially unsure or unwilling to be pubertally staged, it is helpful to offer the family some time outside the consulting room to discuss the matter while the clinician sees the next patient. If necessary, examination can be rescheduled for when an appropriate member of staff is able to attend. Self‐assessment for pubertal staging has been recommended as an alternative option but we advise against making clinical decisions based on this. In those rare instances where a patient refuses examination by any member of staff, measurement of gonadotrophins and sex steroids will help to indicate if puberty has started. Bone Age Pelvic Ultrasound Findings should be compared with normative data (e.g. Griffin et al. 1995). This is required where there is diagnostic uncertainty and/or when treatment is contemplated. The investigation of puberty may include the following tests: Normative data are shown in Table 5.3. Table 5.3 Normative data for LH and FSH before (basal) and after (peak) stimulation with 100 μg intravenous LHRH, derived for 85 subjects aged 3–17 years with no evidence of endocrine disease. Reference range for testosterone and estradiol is from the Institute of Biochemistry, Glasgow Royal Infirmary. a To convert testosterone to ng/d and estradiol to pg/m used these conversion factors: testosterone nmol × 28.8 = ng/dL; estradiol pmol/L divide by 3.671 = pg/ml. Sexual precocity: A general term meaning early sexual development of any kind, with no aetiology implied (Table 5.4). True central precocious puberty ( CPP ): CPP is defined as normal puberty, resulting from activation of the hypothalamus, and following a normal sequence, but occurring abnormally early. In the United Kingdom, the age cut‐offs are before 8 years in girls and before 9 years in boys. Any such definition should take account of population norms and demographic change. A study from the United States Pediatric Research in Office Settings network highlighted concern that standard cut‐offs for defining precious puberty were outdated and that evaluation of either breast development or pubic hair was indicated, should this occur in white girls aged <7 years and black girls aged <6 years (Kaplowitz and Oberfield 1999). In the United Kingdom, age at menarche has changed little but there is evidence that onset of breast development is earlier. A study by O’Connor showed that age at menarche decreased by 0.30 years between 1948 and 2005, while age at onset of breast development decreased by one year (O’Connor 2013). Since assessment of breast development can be difficult in overweight and obese girls, caution is needed in interpreting data which suggests an increase in the prevalence of female early/precocious puberty. Precocious pseudopuberty: Sexual precocity caused by the abnormal secretion of sex steroids independent of hypothalamo–pituitary control. Thelarche: Isolated breast development, common in infants and preschool children, in the absence of other symptoms and signs of sexual precocity. Thelarche variant: This term was first used by Richard Stanhope and Charles Brook from London to describe girls in whom thelarche is persistent or slowly progressive, often associated with a moderate increase in height velocity and bone age, and sometimes vaginal bleeding, but the GnRH test shows a prepubertal response (Stanhope and Brook 1990). Exaggerated adrenarche: In some individuals, adrenarche, a term which refers to the activation of adrenal androgen production normally occurring between six and eight years is associated with sufficient androgen secretion to cause symptoms and signs of sexual precocity. This phenomenon is often mistakenly referred to as ‘premature adrenarche’ – a term which should be confined to adrenarche occurring before six years of age. Premature pubarche: A descriptive term simply meaning early onset of pubic hair development which usually occurs in the context of exaggerated adrenarche. Premature menarche: Cyclical uterine bleeding (confirmed by identifying an endometrial echo on pelvic ultrasound at the time of vaginal bleeding) in the absence of other symptoms and signs of sexual precocity. Tables 5.4 and 5.5 show the types and causes of sexual precocity in boys and girls. The data illustrate that: Table 5.4 Types of sexual precocity encountered at Royal Hospital for Sick Children, Glasgow, 1989–2009. a Caused by: feminizing adrenal adenoma (1), feminizing ovarian tumour (1), virilizing adrenal adenoma (1), virilizing ovarian tumour (1), McCune–Albright syndrome (2), simple virilizing 21‐hydroxylase deficiency (1), 11‐hydroxylase deficiency (1), cause unknown (2). b Simple virilizing 21‐hydroxylase deficiency (3), gonadotropin‐independent precocious puberty (testotoxicosis) (1), virilizing adrenal adenoma (2), Oxymetholone therapy for aplastic anaemia(2), iatrogenic (accidental absorption of paternal testosterone gel) (1) Table 5.5 Aetiology of true central precocious puberty (onset ≤8 years in girls and ≤9 years in boys) in 189 patients seen at RHSC, Glasgow, 1989–2009. Abbreviations: ALL, acute lymphoblastic leukaemia; NF, neurofibromatosis; NHL, non‐Hodgkin’s lymphoma; 11‐OHD, 11‐hydroxylase deficiency; SV21‐OHD, simple virilizing 21‐hydroxylase deficiency. The following two key aspects must be addressed: Androgen‐Related Oestrogen‐Related NB Mood swings and behaviour changes are important in deciding whether or not to treat precocious puberty, but must be gauged carefully, and where possible, separated as far as possible from ‘normal’ difficult behaviour. Features Suggesting the Cause of Sexual Precocity Physical Assessment Figure 5.3 Precocious puberty with B4 development in a seven‐year‐old girl with neurofibromatosis and optic nerve glioma. In a straightforward case, there will be a history of breast development followed by other features of normal puberty including an increase in growth rate, development of pubic and axillary hair, vaginal discharge, mood swings, and sometimes vaginal bleeding (Table 5.5). Examination will show a girl who looks older than her chronological age, with a height centile above the parental target range and features of normal puberty. Magnetic resonance imaging (MRI) focusing on the hypothalamic–pituitary area and looking for a tumour or hamartoma is the investigation of choice, since resolution is better than with computed tomography (CT) scanning and MRI does not expose the child to ionizing radiation. The likelihood of finding a tumour or hamartoma in girls with an onset of puberty between the ages of six to eight years who are otherwise healthy with no neurological symptoms or signs was about 2% in a French study (Chalumeau et al. 2002). Therefore, an MRI may be unnecessary in this group depending on the clinical situation. The younger the child, the greater the chance of finding a cranial lesion with an incidence of 20% when the onset of puberty is <6 years. The object of treatment is to prevent early epiphyseal closure with compromise in final height, and to alleviate psychosocial problems in the child and family. In some cases, the pubertal manifestations will remain static or even regress and no treatment will be necessary. In other cases, because the child’s age is borderline (i.e. seven to eight years) and the family is coping well or because the progress of puberty is very slow, treatment may not be indicated. Some families do not wish to have treatment that they perceive as interfering with the natural course of events, especially if the CPP is familial. A withdrawal bleed can occur in the first four weeks following the start of treatment due to an initial agonist effect of GnRH analogue and by the fall in the oestrogen levels. Provided that families are warned of this possibility, there is usually no need to give pre‐emptive treatment. However, in selected cases, patients can be treated with cyproterone acetate, starting three days before and continuing for the first two weeks after giving GnRH analogue. Occasionally treatment can be associated with headaches and menopausal symptoms such as hot flushes. Local problems such as erythema and sterile abscesses at the injection site may also sometimes occur. Rarely, the CPP is secondary to a tumour in the hypothalamic–pituitary area. In such cases, the histology determines the prognosis. Gliomas tend to be more aggressive than astrocytomas whereas hamartomas are benign. Treatment of the causal lesion usually has no effect on the course of puberty. Hypothalamic hamartomas should not be removed surgically to treat the precocious puberty since this can be treated with GnRH analogues. However, hamartomas causing mass effect or gelastic seizures (epileptic fits characterized by laughing) may require surgical management. Treatment can be expected to prevent or stop menses, to improve mood swings, and to decrease height velocity. Usually, treatment maintains the patient at the same pubertal stage as at diagnosis although occasionally there may be a small reduction in breast size. The parents should be told that treatment does not reverse adrenarche so that pubic hair may continue to develop. In a minority of girls, puberty continues to progress and in those the monthly preparation of GnRH analogue can be given three‐weekly, or the three‐monthly preparation can be given 9–10‐weekly. The child should initially be seen three‐monthly for clinical assessment and auxology, and then six‐monthly once the symptoms have largely settled and the tempo of puberty is clear. Some centres perform regular pelvic ultrasounds and gonadotrophin measurement but we believe that clinical symptoms, breast development stage, and growth rate are better reflections of pubertal status. Bone age should be performed annually. In girls with very slowly progressive CPP, there will be little impact on final height. Many girls with untreated CPP reach a final adult height within the adult reference range. However, these heights may be towards the lower end of the reference range with final heights of 151–155 cm being not uncommon. Final height in treated girls with CPP will depend on when puberty started and when treatment was commenced. Data on final height suggests that treatment does not fully recover lost height potential and children often do not attain their mid‐parental height, possibly because the growth spurt suppressed by treatment does not resume when treatment is stopped. In one study of girls treated till an average age of 11 years, the mean adult height was 160 cm. The timing of stopping GnRH therapy should be discussed with the family. We have found that menses occurs approximately 12–18 months after discontinuing the injections. This has led us to recommend stopping therapy in the middle to the end of the last year of primary school so that menses usually start during the first year of secondary school. Families whose girls have learning disabilities may wish to continue treatment for longer but suppressive therapy beyond 11–12 years of age carries the theoretical risk of preventing normal calcium accretion by the skeleton, and so we are reluctant to continue beyond this age. A large cohort study from the US has shown an important link between early menarche and subsequent problems with depression during and beyond adolescence, and anti‐social behaviour (Mendle et al. 2018). The authors of this study highlighted the importance of follow‐up beyond the paediatric age range. It is evident that the mental health of girls with CPP has tended to be overlooked in the past, with too much focus on diagnosis and medical treatment. Awareness of the psychological vulnerability of these girls is important in pre‐empting and managing emotional problems. The diagnosis is suspected when there is progressive feminizing precocity associated with a prepubertal GnRH test or more often undetectable LH and FSH throughout the test (see Table 5.4). Causes include adrenal adenoma, granulosa cell tumour of the ovary, and McCune–Albright syndrome. The last disorder may present with breast development and vaginal bleeding in a girl with café au lait patches and bony symptoms or signs on X‐ray. Rarely, severe and protracted primary hypothyroidism results in cross‐reaction between elevated thyroid‐stimulating hormone (TSH) levels and gonadotrophin receptors, leading to breast development and vaginal bleeding (Van Wyk and Grumbach 1960). Serial imaging of the ovaries by pelvic ultrasound, adrenal ultrasound, and CT, urine steroid analysis in selected cases, GnRH test, measurement of the tumour markers – α foeto‐protein and β hCG – and a skeletal survey may be required. Surgery is the treatment of choice for adrenal and ovarian tumours. Medical treatment for precocious pseudopuberty is directed at restricting or antagonizing sex steroid production. Useful agents include the following: Secondary true puberty resulting from priming of the hypothalamus will require additional GnRH therapy. NB: Experience with these agents in the management of sexual precocity is necessarily limited because of the rarity of the conditions concerned. These medications should therefore be prescribed only by specialist centres. Premature thelarche is usually seen in preschool girls (Figure 5.4). Sometimes the breast development has been present from birth. More commonly, the mother notices breast development, often asymmetrical and with a tendency to wax and wane, from the age of 6–12 months. There are no other features of sexual precocity and height velocity is normal. The bone age and pelvic ultrasound are consistent with chronological age. Figure 5.4 Premature thelarche with Tanner stage 3 breast development. Bone age and pelvic ultrasound may be waived in mild cases of premature thelarche. In more florid cases, these together with an GnRH test should be performed. The FSH response is often pronounced, with 30‐ or 60‐min. values of up to 25 units/L while LH values are below 4 units/L. Plasma oestradiol is usually unrecordable using standard assays. None is required. Three‐ or four‐monthly clinic visits over a one‐year period to confirm that breast development is static or regressing. Girls with thelarche variant are usually between five and eight years of age and present with breast development which persists and may progress slightly in association with slight increase in height velocity and modest advance in bone age. Vaginal bleeding sometimes occurs. Pelvic ultrasound may show an oestrogen effect on the uterus, but is less marked than for CPP. Ovaries may show some enlargement with an increase in follicular activity. Thelarche variant is diagnosed in the context of persistent but mildly progressive sexual precocity in conjunction with a prepubertal GnRH test, modest bone age advance and uterine development, normal imaging of the adrenal glands, and an indolent clinical course. Thelarche variant probably includes a variety of conditions including CPP that is too mild to be detected on the GnRH test, and subtle alterations in responsiveness of the ovaries to normal prepubertal LH and FSH levels. Usually, no treatment is required but the girls must remain under surveillance and undergo serial auxology, pubertal staging, and pelvic ultrasound examination. Rarely, girls may show unacceptable progression of symptoms and signs so that treatment is requested. Under these circumstances, specialist referral is recommended. Peripheral antagonists, such as cyproterone, may be tried. This condition occurs in girls usually between four and eight years of age in whom cyclical vaginal bleeding occurs in the absence of other features of sexual precocity. The diagnosis is made by identifying an endometrial echo on pelvic ultrasound when the child is experiencing vaginal bleeding. It is essential not to diagnose premature menarche unless the above criteria are satisfied and other causes of vaginal bleeding have been excluded. These include recurrent vulvovaginitis, foreign body, child sexual abuse, other causes of trauma, and vaginal tumours (e.g. rhabdomyosarcoma). Pelvic ultrasound examination is usually sufficient but an GnRH test may be required if there is any suspicion of symptoms other than vaginal bleeding. No hormone treatment required. Three‐ to four‐monthly review to confirm normal auxology and no development of other features of sexual precocity. This is by far the most common cause of androgenicity in girls, and presents from six years of age with a history of body odour, greasy skin and hair, weight gain, and sometimes mood disturbance, usually in association with some pubic and/or axillary hair development. The distribution of the pubic hair is characteristically over the labia majora rather than the mons pubis. Adrenarche must be distinguished from simple virilizing and non‐classical 21‐hydroxylase deficiency, and androgen‐secreting tumours of the adrenal glands or ovaries. This is usually easy on clinical grounds as the symptoms and signs of adrenarche are relatively mild and in particular there is no clitoromegaly. Bone age may be advanced by one year, sometimes more if obesity is an accompanying feature. In all but the mildest cases, blood should be taken for serum testosterone, androstenedione, 17‐OHP and DHEAS (see Chapter 8 for normal values), preferably at 8 a.m. to detect mild 21‐hydroxylase deficiency. If baseline androgens are elevated or clinical features are particularly marked, then further investigation with a standard Synacthen test and adrenal imaging are indicated to exclude non‐classical congenital adrenal hyperplasia (CAH) or an adrenal tumour. No treatment is required. If the serum androgens show a mild elevation, particularly of androstenedione and DHEAS, then one further clinic visit in four to six months to confirm normal growth rate and lack of progressive virilization is sufficient before discharge. Although a link between exaggerated adrenarche and low birth weight/smallness‐for‐gestational age and subsequent hyperandrogenism (e.g. polycystic ovary syndrome [PCOS]) has been described (Ibáñez et al. 1998), the majority of girls examined in Scotland were of normal birth weight and clinical features of androgen excess were mild (Paterson et al. 2010). We do not normally, therefore, recommend long‐term follow‐up in these patients. Simple virilizing 21‐hydroxylase deficiency is suggested by the combination of advanced growth, clitoromegaly, pubic/axillary hair development and bone age advance, especially if the girl is younger than six years (see Table 5.4). Virilizing tumours of the adrenal or ovary are suggested by a shorter history, so that features such as tall stature have not usually had time to become manifest (Figure 5.5). Figure 5.5 Virilizing ovarian tumour (outline shaded on skin) causing pubic hair and clitoromegaly in a three‐year‐old girl. Measurement of serum testosterone, 17‐OHP, androstenedione, and DHEAS under basal conditions and in selected cases after stimulation with Synacthen will demonstrate any abnormalities in serum androgens. Urine steroid analysis and/or molecular studies may be required to help identify an adrenal enzyme defect. Adrenal MRI or CT may be required as well as careful evaluation of the ovaries on ultrasound. If, despite imaging, a tumour cannot be identified, then selective venous sampling from the adrenal veins should be carried out at a specialist centre. Simple virilizing 21‐hydroxylase deficiency is discussed in Chapter 8. Virilizing tumours of the adrenal glands and ovaries are treated surgically. This involves three‐ to four‐monthly assessment to oversee regression of the features of sexual precocity and a normal growth rate. Depending on availability, MRI (preferable), CT scanning or ultrasound scanning of the adrenals or ovaries should be performed annually. Long‐term follow‐up is needed to pre‐empt the development of CPP because of priming. This condition is rare in boys, and its occurrence must prompt a search for an underlying cause (Figure 5.6) (see Table 5.5). The prevalence of a brain tumour in boys is 40–90%. Clinical symptoms include rapid growth rate, behaviour disturbances, a deepening of the voice, and enlargement of the genitalia. Examination will demonstrate symmetrical enlargement of the testes to volumes ≥4 ml. Figure 5.6 Hypothalamic hamartoma in a six‐year‐old boy, causing gelastic seizures and central precocious puberty. The combination of sexual precocity with bilateral testicular enlargement makes the diagnosis of CPP in boys easy. If the testes are prepubertal in volume and/or asymmetrical, the causes of precocious pseudopuberty must be sought (see section ‘Precocious pseudopuberty’). A GnRH test to confirm true puberty, with measurement of serum testosterone, followed by imaging of the hypothalamic–pituitary area by MRI. The tumour markers – β hCG and α foetoprotein should be measured to detect germinomas. Treatment is with a GnRH analogue as described for girls with CPP. Any underlying cause should be treated. This is as for CPP in girls. In cases where no brain tumour is found on initial imaging, serial imaging at 6–12‐monthly intervals for a 2–3‐year period is required to detect an evolving lesion. Testicular size can be expected to diminish and genital development may show some regression. There should also be a reduction in any aggressive behaviour and in the number of erections. Simple virilizing 21‐hydroxylase deficiency will result in excessive androgen secretion from the adrenal glands (see Table 5.4). Adrenal tumours may be androgen‐secreting. In the testes an activating LH receptor mutation affecting all the Leydig cells (germline) causes a condition known as gonadotropin‐independent precocious puberty (‘testotoxicosis’). An activating LH receptor mutation affects only a single cell (somatic) and results in a Leydig cell tumour (Figure 5.7). Precocious pseudopuberty may be seen in boys receiving the anabolic steroid Oxymetholone which exerts a direct androgenic effect (as well as priming the hypothalamus and causing true precocious puberty). Figure 5.7 Leydig cell tumour causing slight enlargement of left testis with contralateral atrophy, and precocious pseudopuberty in a five‐year‐old boy. Common to all these rare conditions are testes that are either prepubertal in volume (in the case of adrenal disorders or an exogenous cause), modestly and symmetrically enlarged (i.e. 3–4 ml in testotoxicosis and hCG‐secreting tumours), or unilaterally enlarged with contralateral atrophy (in Leydig cell tumours). By contrast, in CPP, both testes are at least 4 ml in volume. The GnRH test will show suppressed LH and FSH values in precocious pseudopuberty unless the primary disorder has activated the hypothalamus. Investigations of the underlying cause may include β‐hCG and α‐foetoprotein measurement; serum testosterone, 17‐OHP, androstenedione, and DHEAS before and, in selected cases, following Synacthen stimulation; urine steroid analysis; testicular ultrasound; and adrenal MRI or CT. Surgery is the treatment of choice for tumours. Medical treatments include Ketoconazole, Cyproterone, Testolactone, and Flutamide. Adrenarche is less common in boys than girls, but is still the most common cause of androgenicity. The clinical diagnosis, investigations, and management are as for girls. Feminizing tumours of the adrenal glands and testes are extremely rare. Gynaecomastia, caused either by enhanced oestrogen production from aromatization of testosterone at puberty, or by increased tissue sensitivity to normal circulating oestrogen levels, is a relatively common problem in boys and is discussed below. Delayed puberty: This is arbitrarily defined as absence of signs of secondary sexual development in a girl aged 13 years or a boy aged 14 years. A more practical and helpful definition is delay in the onset, progression or completion of puberty sufficient to cause concern to the adolescent, parents, or physician. Absent or arrested puberty: Puberty that either does not begin, or having begun, does not progress (in which case the term ‘mid‐pubertal arrest’ is often used). Delayed menarche: First period after 15 years. Primary amenorrhoea: Failure of periods to begin. Secondary amenorrhoea: Cessation of established periods. Oligomenorrhoea: Infrequent periods (e.g. occurring > every 35 days or < 4–9 per year). Delayed puberty be classified simply as central or peripheral, depending on whether the site of the problem lies in the hypothalamo–pituitary axis or in the gonads. Central delayed puberty can be further subdivided into delay with intact hypothalamo–pituitary axis, and delay caused by impairment of the hypothalamo–pituitary axis. Table 5.6 gives a working classification of delayed puberty and its causes. Table 5.6 Classification of delayed and abnormal puberty (including delayed/absent menarche). Abbreviations: CAIS, complete androgen insensitivity syndrome; CDGA, constitutional delay in growth and adolescence; FSH, follicle‐stimulating hormone; GnRH, gonadotrophin‐releasing hormone; H–P, hypothalamo–pituitary; LH, luteinizing hormone. Almost all boys and most girls with delayed puberty have simple constitutional delay in growth and adolescence (CDGA). In pathological delayed puberty, the cause may be obvious from the past medical history. The challenge is to diagnose the occasional case of pathological delay in puberty in which the cause is not evident, and the more complicated cases with a multifactorial aetiology (e.g. constitutional, nutritional, social). (If indicated.) By far the most common cause of delayed puberty, CDGA is usually easily diagnosed by the history of long‐standing short/borderline short stature during childhood. Following secondary school entry at 11 years, there is a decline in height status so that the boy becomes relatively shorter for age. Commonly, there is a family history of delayed puberty. Frequently, the boy expresses distress to his parents, sometimes related to teasing, but more often because of discomfort at being small and young‐looking for age. There is frequently embarrassment about taking showers at school. In boys of 13 years and over, the testes usually show some enlargement (4 ml), but no penile enlargement (i.e. Tanner stage G2). CDGA should usually be a positive diagnosis and not one of exclusion. However, the presence of possible contributory factors such as poor nutrition, socio‐economic and psychological difficulties, and chronic ill health (e.g. severe asthma) can render assessment difficult. Endocrine disturbance is suggested when the boy is not short or borderline‐short; when the short stature and delayed puberty are out of context with the family pattern; when the testes are undescended or abnormal; when there are symptoms and signs such as headache or optic atrophy suggestive of craniopharyngioma; or when penis and testes are hypoplastic, especially in association with impaired sense of smell (Kallmann syndrome). None is necessary in CDGA except for bone age estimation which usually shows delay. The boy should be reassured that he is normal, simply a late developer. Prediction of adult height is helpful, providing that a single experienced observer performs the bone age assessment. The following medical treatments can be offered to enhance growth rate and expedite the features of puberty without affecting final height outcome: Six‐monthly visits to the clinic may be helpful in reassuring untreated boys. Boys treated with Oxandrolone or testosterone should be seen six‐monthly for two visits following treatment to ensure satisfactory progress. Occasionally, a repeat course of testosterone is indicated. If there is any doubt about the diagnosis of CDGA, it is prudent to keep the boy under observation until testicular volumes are 10 ml. CDGA in girls is less common than in boys but remains the most common cause of delayed puberty (Figure 5.8). Characteristically, the tempo of puberty, when it starts, is slower and a gradual increase in height rather than a discernible adolescent growth spurt is observed (Figure 5.8). Figure 5.8 Growth chart of girl with delay in growth and adolescence showing attenuated and late growth spurt with menarche at 15 years of age. Final height is equal to the mid‐parental height (MPH). M, menarche. CDGA in girls must be distinguished from Turner’s syndrome as well as other pathological causes of delayed puberty. In doubtful cases, the chromosomes and basal gonadotrophins should be checked and a pelvic ultrasound performed. It should be noted that failure to identify one, and occasionally both, ovaries may occur in normal girls. In contrast to boys, who can be given a sizeable dose of testosterone with no ill effect on final height, oestrogen therapy in girls is both less effective and more likely to cause premature epiphyseal closure. Occasionally, pubertal delay is so marked and growth rate so slow that there is a need for treatment. In these circumstances, GH levels should be measured. Daily ethinyloestradiol 2 μg for a 1–2‐year period can be given, with concurrent GH administration if stimulation testing shows GH insufficiency. Sometimes the chronic disease is obvious, and the child is referred simply to confirm that no other pathology is responsible and to advise as to possible treatment. The clinician must assess the severity of the chronic disease and confirm that the severity of the delay in growth and adolescence matches the severity of the chronic condition. If this is not the case, then further investigation is indicated. A small proportion of girls and boys presenting with delayed puberty will be found to have chronic disease as the underlying cause. Gastrointestinal conditions, such as coeliac disease and Crohn’s disease, can be notoriously silent. The following screening investigations are of value in selected cases: Some adolescents have clear evidence of damage to the hypothalamo–pituitary axis (e.g. craniopharyngioma) and will clearly not enter or complete puberty without treatment (see Table 5.6). Adolescents with delayed puberty who do not fulfil the criteria of CDGA pose some difficulty, for the GnRH test cannot distinguish between physiological central delay and true GnRH or gonadotrophin deficiency. A history of impaired sense of smell, testicular maldescent and/or surgery and assessment of academic performance must be sought. Examination includes a search for dysmorphic features, systemic examination, and detailed examination of the external genitalia. Investigations are detailed above. In doubtful cases, it is best to induce puberty with testosterone from the age of 13–14 years onwards until either full secondary sexual development has occurred or spontaneous testicular development to 10 ml has taken place (indicating intact endogenous gonadotrophin secretion). If spontaneous testicular enlargement has not occurred by the age of 16–18 years, replacement therapy should be discontinued and the gonadal axis fully re‐evaluated. Table 5.6 classifies delayed or abnormal puberty. Primary hypogonadism is suggested by the combination of: These will show elevated FSH (above 10 units/L). An hCG test may show impaired testosterone rise, or no rise in severe cases. Anorchic subjects will clearly require therapy, as will individuals with arrested secondary sexual development, particularly if testosterone levels are low with poor response to hCG. By contrast, boys with low testicular volumes but normal genital and pubic hair development and acceleration of linear growth will not require treatment. As with central hypogonadism, it is best to treat doubtful cases pre‐emptively rather than waiting, which carries the risk that development may not progress satisfactorily, resulting in distress to the patient. The available treatments for delayed puberty and hypogonadism in males in the United Kingdom are summarized in the British Society for Paediatric Endocrinology and Diabetes guidelines (El‐Khairi et al. 2016). Intramuscular preparations include: The oral preparation is: Other preparations include: The following regimen will bring about satisfactory secondary development in hypogonadal boys over three years: Nebido 1 g intramuscularly every 12 weeks, modifying the dose according to clinical response, is an effective treatment for maintaining secondary sexual development in hypogonadal males. The injection is given by the family practitioner or nurse and renders compliance/adherence less of a problem than with oral replacement. The large 4 ml volume of the preparation may be best given as 2 ml into each buttock rather than a single injection and some adults prefer monthly Sustanon 250 mg rather than the larger three‐monthly injection. An alternative to intramuscular testosterone is oral testosterone undecanoate 40 mg per day for the first year, 80 mg per day for the second year and 120 mg per day thereafter. The causes of hypothalamic and pituitary gonadotrophin deficiency in girls are similar to boys, although isolated GnRH and gonadotrophin deficiency are rarer. Investigations will show low LH and FSH levels and management is as for boys. Gonadal dysgenesis as a result of Turner syndrome or iatrogenic damage from total body irradiation are the main causes of primary gonadal failure in girls. Basal FSH and LH will be elevated in frank ovarian failure, with FSH values of ≥ 10 u/L indicating significant impairment. A GnRH test may be required to demonstrate milder germ cell damage. Anti‐Müllerian hormone (AMH) is secreted by the granulosa cells of the ovary in girls and serves as an index of ovarian reserve (Visser et al. 2013), values of <0.05 ng ml (<0.36 pmol/L) indicating absence of ovarian tissue. Pelvic ultrasound may show a less‐developed uterus than would be expected from the pubertal stage, while ovaries may be small or unidentifiable. In doubtful cases, pubertal induction should be offered from 12 to 13 years. It is important for oestrogen replacement to be gradual, not only to avoid premature fusion of the epiphyses, but also to prevent unsightly overdevelopment of the areolae of the breast. Until recently the three‐year oral induction regime most commonly used in the United Kingdom used Ethinyl estradiol, giving 2 μg per day for the first year, 4 μg per day for the second year and then 6, 8 and 10 μg per day during the third year, in four‐monthly steps. Recent consensus statements have recommended a more physiological approach with transdermal rather than oral oestrogen for pubertal induction (Gravholt et al. 2017) and the use of natural 17β‐estradiol rather than synthetic oestrogens (Matthews et al. 2017). The oral and transdermal regimens proposed by Matthews et al. are given in Tables 5.7 and 5.8. Table 5.7 Regimen for pubertal induction using 25 μg/24 hours of 17β‐estradiol matrix patches applied once or twice weekly and left in situ for three to four days. Source: Reproduced from Matthews et al. (2017) with kind permission from Archives of Disease in Childhood. Table 5.8 Regimen for pubertal induction using 1 mg 17β‐estradiol tablets. Source: Reproduced from Matthews et al. (2017) with kind permission from Archives of Disease in Childhood. Progesterone is usually introduced in the final 12 months of induction. It is important not to start this treatment too soon, in order to maximize the time for uterine and breast development. When breakthrough bleeding occurs, therefore, a pelvic ultrasound examination should be carried out. If the uterus is under‐developed with a thin endometrium, then progesterone treatment should be postponed; but started if adequate uterine development with a thick endometrium is demonstrated. Preparations include Norethisterone 5 mg, Utrogestan 200 mg, and medroxyprogesterone acetate 5 mg, all given orally once daily for the first 12 days of each calendar month. Following successful pubertal induction, maintenance treatment is required. Administration of a low‐dose combined contraceptive pill, e.g. Loestrin 20 (21 days on and 7 days off) is convenient but has the disadvantage of rendering the patient untreated with oestrogen for three months of the years. A more physiological approach is to administer either oral 17β‐estradiol 2–3 mg daily or twice‐weekly transdermal 17β‐estradiol for days 1–25 of each calendar month, adding in a progesterone preparation for days 12–14, to mimic the natural cycle (Christin‐Maître 2017). Post induction oestrogen replacement should always be given in consultation with a gynaecology, reproductive endocrine, or adult endocrine colleague. It is very important for late adolescent/early adult patients to be carefully counselled concerning the benefits and the importance of good adherence to hormone replacement, since this affects not only sexual but bone, cardiac, and mental health. Prolactin‐secreting pituitary adenomas are rare in childhood and adolescents. They are classified according to size into microadenomas (≤10 mm diameter) and macroadenomas (>10 mm diameter). Clinical features may be endocrine‐related, the high prolactin levels causing galactorrhoea, gynaecomastia, and primary or secondary amenorrhoea; related to tumour mass with headache and visual field defect; or to both hormonal and mass effects. Prolactin levels are usually markedly above the reference range of <440 mIU/L (<25μg/L) in women and <350 mIU/L (<20μg/L) in men; a study of 26 children and adolescents showed median (range) prolactin 94 (70–500) μg/L in 11 patients with microadenoma and 720 (145–3300) μg/L in 15 with macroadenoma (Colao et al. 1998). Milder hyperprolactinaemia may be associated with stress and with anti‐psychotic medication. Treatment of prolactinoma is with the dopamine agonist Cabergoline, starting at 0.25 mg weekly and increasing depending on the response of prolactin levels and tumour size, is now the treatment of choice. Bromocriptine 1.25 mg nightly increasing to 2.5 mg twice daily can also be used. The usual shrinkage of tumours and excellent biochemical control with these agents means that other treatments such as surgery and radiotherapy are rarely needed and reserved for refractory cases. Patients should be managed jointly with an experienced adult endocrinologist. The most common complaint is that of irregular, prolonged, and heavy uterine bleeding, sometimes associated with considerable discomfort. This is related to immaturity of the hypothalamic–pituitary–ovarian axis and to anovulatory cycles after onset of the menses, and should be considered a normal phenomenon during the first two years following menarche. Maintaining a menstrual diary will help assess the severity of the problem. Usually, no treatment is required other than simple analgesia such as mefenamic acid. The antifibrinolytic drug, tranexamic acid, can also be used during episodes of bleeding to diminish blood loss. However, the symptoms may be severe and affect schooling and further treatment may be required. The following strategies may help: Prior to prescribing the above two medications, hypertension and a family history of deep vein thrombosis should be excluded. Caution should also be exercised in patients with epilepsy and liver dysfunction. In cases of severe and frequent dysfunctional uterine bleeding and when bleeding is so heavy that it is accompanied by clots, one should consider undiagnosed underlying causes such as von Willebrand’s disease. An FBC to exclude anaemia and thrombocytopaenia, and iron and coagulation studies may be required. Dietary advice and iron supplements may also be required. If there is no improvement in the dysmenorrhoea, alternative diagnoses such as endometriosis or obstruction of the lower genital tract should be considered and the patient should be seen by a gynaecologist. Amenorrhoea and oligomenorrhoea are commonly seen as part of normal delayed puberty, and are particularly likely to occur in the context of a low body mass caused by extreme physical training, poor nutrition, and eating disorders, such as anorexia nervosa. It is rare for girls to start their periods if their body weight is <40 kg. A minimum of three to four periods a year are required to ensure adequate oestrogenization and to minimize the risk of osteoporosis. If the patient has a completely normal physical examination with secondary amenorrhoea, consider administering medroxyprogesterone, 10 mg once daily for 5 to 10 days to diagnose anovulation as the cause of the amenorrhoea and to see if a withdrawal bleed will occur (progesterone challenge test). If bleeding occurs, then adequate levels of endogenous oestrogen and a normal anatomy can be inferred. Primary amenorrhoea is seen in girls with absent puberty, anatomical disorders of the reproductive tract (e.g. imperforate vagina or Rokitansky syndrome, in which there is congenital absence of the uterus and some or all of the vagina) and in the syndrome of complete androgen insensitivity (see Chapter 7). None is required where the history and examination indicate the diagnosis. Pelvic ultrasound assessment and blood samples for chromosomes, gonadotrophins, and sex steroids are helpful in selected cases. There is an overlap between hirsutism (inappropriate/excessive hair growth in androgen‐dependent sites such as moustache and beard area, chest, etc.) and hypertrichosis (generalized increase in body hair). Hirsutism may be seen in the following instances: Hypertrichosis may be seen in the following instances: Androgen excess can be gauged by accompanying features such as tall stature, bone age advance, clitoromegaly, etc. PCOS is suggested by accompanying oligomenorrhoea and/or obesity. These are unnecessary if the hirsutism is mild and the cause obvious (e.g. racial). In selected cases, investigations should be performed as for PCOS. Treatment is directed at the underlying cause, and where this is not possible, cosmetic strategies include bleaching of facial hair, removal of hair by electrolysis, and referral to a dermatologist for consideration of laser treatment. Endocrine Society guidelines from the USA continue to support the diagnosis of PCOS being made in the presence of two out of three key criteria (Legro et al. 2013). These are: The latter are defined according to the Rotterdam consensus criteria (2004) as at least one ovary with either 12 or more follicles of 2–9 mm diameter and/or a volume of >10 ml in an ovary with no dominant follicle >10 mm, and after exclusion of other causes of androgen excess or related disorders. Ovaries are often bilaterally enlarged with peripheral distribution of the follicles and an increased in stromal density. PCOS may occur in postmenarcheal teenagers and, very occasionally, in premenarcheal girls. The clinical features of PCOS include obesity, hirsutism, greasy skin and hair, acne, oligo/amenorrhoea, and subfertility. Polycystic ovaries are not a prerequisite for the diagnosis of PCOS, and are found in approximately 25% of normal women. Laboratory features include elevated serum androgens (particularly testosterone, free testosterone, and androstenedione), LH hypersecretion (with an LH to FSH ratio of 3 : 1 or more), and hyperinsulinism with low SHBG levels. Frequently, only some of the clinical and laboratory features are present. PCOS can be regarded as a disease complex which is heterogeneous in nature and in which there is no single underlying cause. Insulin resistance and consequent hyperinsulinaemia in association with obesity are commonly seen in women with PCOS but some patients are slim. The frequent association of PCOS with obesity may have a cumulative deleterious effect on glucose homeostasis through hyperinsulinaemia, with an increased prevalence of type 2 diabetes. Ovarian hyperandrogenism results from the action of high insulin levels on the insulin receptor; cross‐reaction with the IGF‐1 receptor; and synergism with LH hypersecretion. Hyperinsulinaemia also decreases the synthesis of SHBG, leading to an increase in circulating free testosterone. Genetic and environmental factors contribute to the development of PCOS, affecting women from South Asia more severely, with an increased risk of type 2 diabetes. In a girl with a history suggestive of PCOS, investigations are directed at confirming the diagnosis and excluding conditions such as non‐classical CAH and other causes of hyperandrogenism. The full investigation of PCOS with the exclusion of CAH consists of: Testosterone levels are elevated in PCOS but levels of >5.0 nmol l−1 are unusual and suggest an androgen‐secreting tumour of the adrenal gland or ovary. An MRI should rule out adrenal pathology and the pelvic ultrasound should have ruled out an ovarian tumour. DHEAS and androstenedione levels are also often elevated. Thyroid dysfunction can lead to amenorrhoea and hirsutism. Mild cases do not require treatment, nor do girls with polycystic ovaries but who are not showing any features of the condition. The cornerstone of non‐medical management in obese individuals is weight reduction (by dietary measures and exercise) which reduces the hyperinsulinism and the risk of type 2 diabetes. Medical treatment is directed at the hyperinsulinaemia and hyperandrogenism. Metformin is used in hyperinsulinaemic girls, especially in the context of obesity. It increases insulin sensitivity and may help to regulate periods. Periods can be regulated with the oral contraceptive pill which will also aid the skin problems. In those with hirsutism, the combination of an oestrogen with an anti‐androgen will help combat signs of androgen excess as well as regulating periods. Dianette which contains the anti‐androgen cyproterone and ethinyloestradiol and the newer combined contraceptive pill Yasmin which contains drosperidone and ethinyloestradiol can help in this regard. Both preparations have been linked with depression and should not be used lightly. It is important to manage PCOS in collaboration with an adult endocrine physician or a gynaecologist experienced in the field. PCOS is associated with psychological morbidity and an impaired quality of life, and referral to a psychologist or psychiatrist may be indicated. Breast problems can be divided into gynaecomastia in boys and either asymmetrical or symmetrical smallness or largeness of breast size in girls. Apparent gynaecomastia results from an unfortunate emphasis in fat distribution towards the breast area, particularly in obese boys. Exaggerated breast development at puberty is the most common cause of true gynaecomastia in boys. This is usually mild, and transient. More severe gynaecomastia is fortunately less common and almost always idiopathic (Figure 5.9). Normal genital and testicular development should be verified. In severe or doubtful cases, chromosomes for Klinefelter syndrome, basal gonadotrophins, and serum oestradiol should be checked. Occasionally, drugs (prescribed, such as spironolactone, or recreational, such as cannabis) will cause gynaecomastia. Figure 5.9 Idiopathic gynaecomastia in an adolescent boy. If gynaecomastia is severe and causing distressing problems (see Figure 5.9), for example, the boy will not participate in physical activities or have showers in front of his peers, then plastic surgery referral is indicated with a view to mammary reduction by either liposuction or subareolar incision and removal of excess tissue. Occasionally, breast size is unacceptably large to the girl and family, in which case referral to a plastic surgeon for consideration of reduction mammoplasty is indicated. Girls with delayed puberty will have smaller breasts than their peers and can be reassured accordingly. Poor breast growth may occur in girls who have received chest irradiation for cancer (e.g. lung metastases). Once optimal breast size has been attained by waiting for endogenous puberty to complete, or by oestrogen administration in girls with gonadal failure, referral for augmentation mammoplasty should be discussed with the family. If there is reluctance to inject foreign material, such as silicone, into the breasts of teenage girls on the part of the plastic surgeon, then reconstructive surgery should be considered. Asymmetrical breast development is very common in the early stages of puberty, particularly in girls with sexual precocity. Occasionally, postpubertal girls present with an unacceptable discrepancy in breast size. Investigation is unhelpful and referral to a plastic surgeon is indicated for either reduction mammoplasty on one side or augmentation mammoplasty on the other.
Puberty
Physiology of normal puberty
In boys
In girls
In both sexes
Physiology of the menstrual cycle
Onset of puberty
Clinical aspects of normal puberty
Pubertal staging
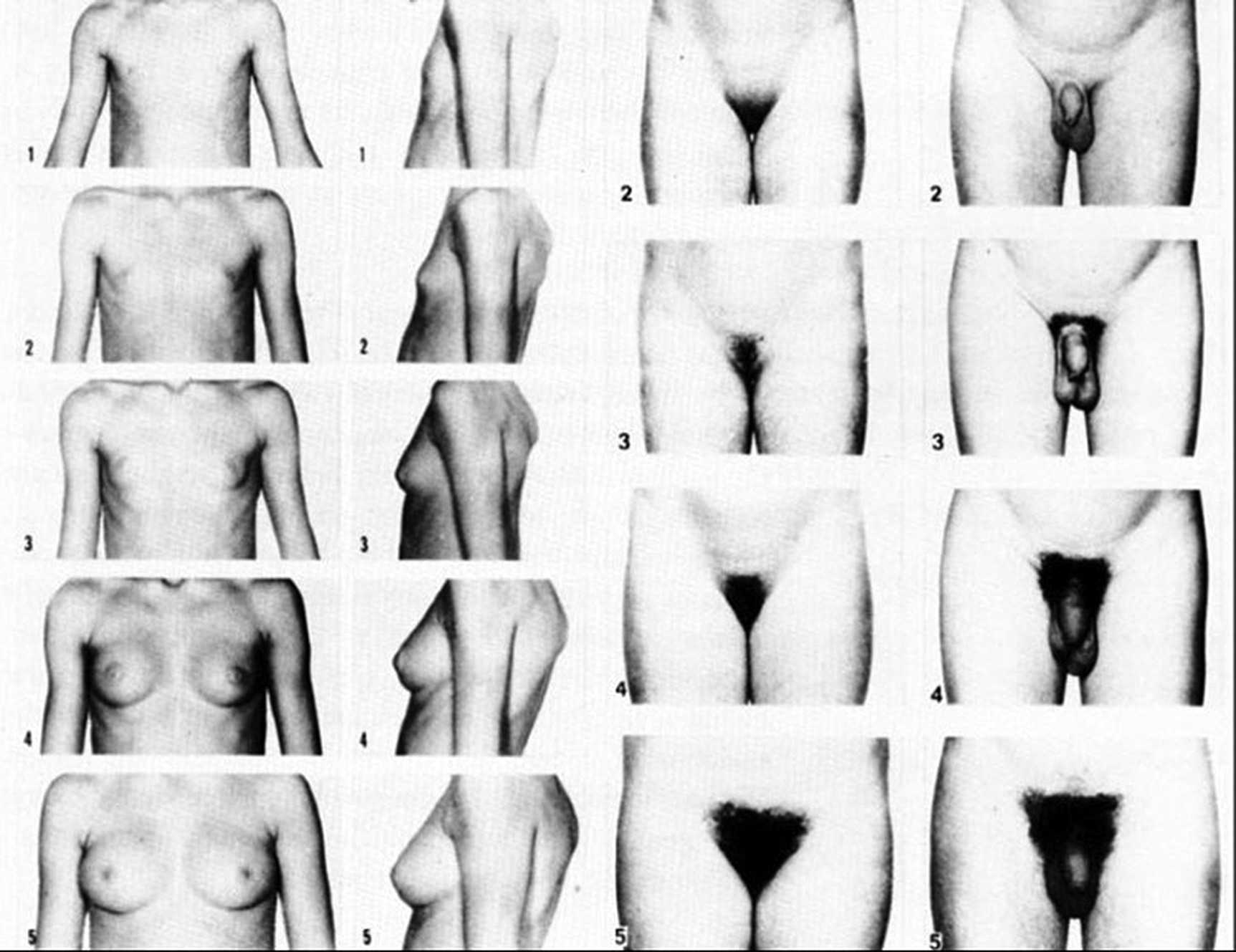
Breast Staging
Genital Staging
Pubic Hair Staging
Axillary Hair Staging
Milestones of puberty
Age (yr)
Height velocity (cm yr−1)
B1
Prepuberty
approx 4–7 from 7 yr
B2
11.2
7
PHV
12.1
8.3 (6.2–10.4).
B3
12.2
8.2
B4
13.1
5
Menarche
13.5
3.6
B5
15.3
?1
Age (yr)
Height velocity (cm year)
G1
Prepuberty
approx. 4–7 from 7 yr
G2
11.6
5
G3
12.9
6.3
G4
13.8
9.3
PHV
14.0
9.5 (7.2–11.7)
G5
14.9
6.2
Pubertal assessment
Clinical Evaluation
Auxology
History and Examination
Diagnostic Imaging
Laboratory Assessment
LH (units l−1)
FSH (units l−1)
Tanner stage
Basal
Peak
Basal
Peak
Testosterone (nmol l−1)a
Estradiol (pmol/l)a
Girls
B1
0.5 (0.5–2.4)
2.4 (0.5–4.9)
2.0 (0.2–5.0)
12.0 (2.2–26.4)
<50
B2
0.55 (0.5–2.6)
9.7 (1.6–16.4)
3.3 (0.5–8.9)
9.9 (6.8–22.2)
B3–4
1.6 (0.5–4.8)
23.6 (6.1–50.6)
5.8 (2.5–7.0)
14.3 (12.0–26.7)
Boys
G1
0.5 (0.2–0.5)
2.0 (0.5–5.11)
1.0 (0.2–4.7)
4.4 (1.0–11.6)
<0.3
G2
1.1 (0.5–2.0)
8.3 (4.4–13.8)
1.7 (0.3–4.2)
2.5 (0.9–11.2)
G3–5
1.9 (1.9–4.0)
16.6 (10.4–21.1)
3.2 (1.8–10.6)
6.3 (2.8–17.0)
Adult
8.7–35.0
180–1500
Sexual precocity
Definitions
Clinical assessment and diagnosis of boys and girls with sexual precocity
Girls
Boys
Central precocious puberty:
Idiopathic
122
5
Secondary
39
15
Precocious pseudopuberty
10a
9b
Exaggerated adrenarche
251
50
Thelarche
104
Thelarche variant
44
Isolated premature menarche
29
Girls
Boys
Idiopathic
122
5
Cranial irradiation
ALL
2
0
Medulloblastoma
1
0
Other tumour
1
0
NHL
0
0
Tumour
Optic nerve glioma (NF)
6 (4)
5 (3)
Craniopharyngioma
1
1
Hypothalamic hamartoma
3
1
Germinoma
0
1
3rd ventricle cyst
1
1
Neurological disorder
Learning disability +/− epilepsy
8
4
Hydrocephalus/spina bifida
11
1
Tuberculous meningitis
1
0
Cerebral palsy
5
0
Priming
11‐OHD
1
0
SV 21‐OHD
1
3
Oxymethalone
0
2
Testotoxicosis
0
1
History
Symptoms related to sex steroid production
Diagnosis and management of sexual precocity in girls
Oestrogen‐mediated sexual precocity
True Central Precocious Puberty
Clinical features
Investigations
Management
Follow‐up
Psychological sequelae of precocious puberty
Precocious Pseudopuberty: Feminizing
Investigations
Treatment
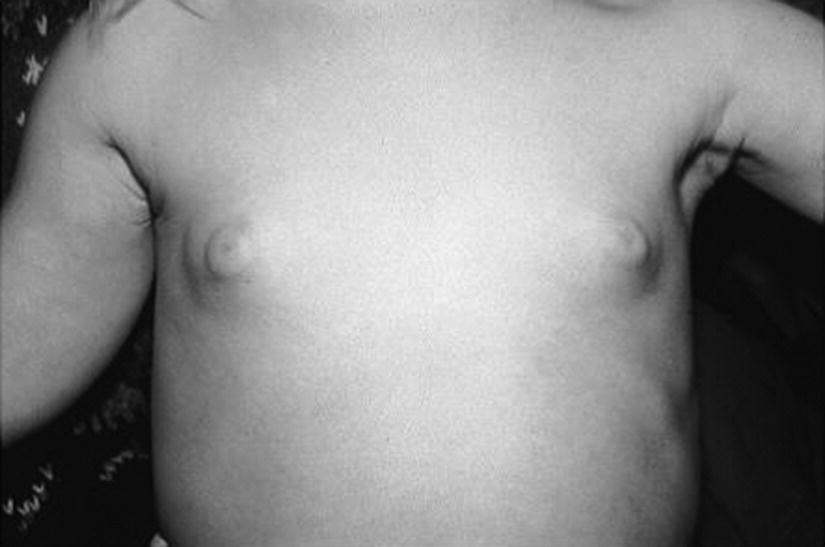
Thelarche
Clinical features
Investigations
Treatment
Follow‐up
Thelarche variant
Diagnosis
Management
Premature Menarche
Diagnosis
Investigations
Treatment
Follow‐up
Androgen‐mediated sexual precocity
Exaggerated Adrenarche
Clinical features
Diagnosis
Investigations
Treatment and follow‐up
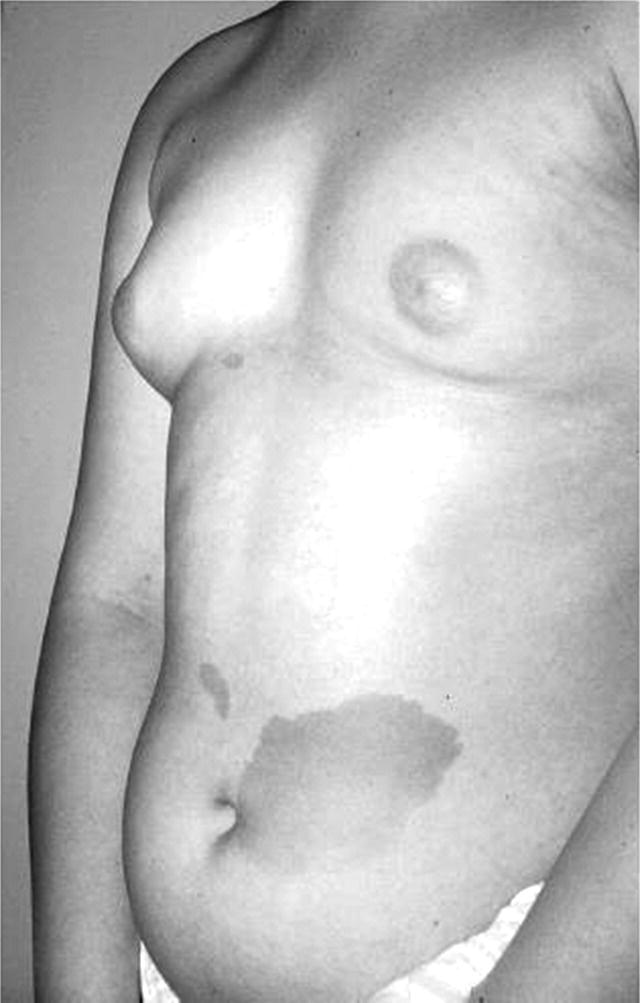
Precocious Pseudopuberty: Virilizing
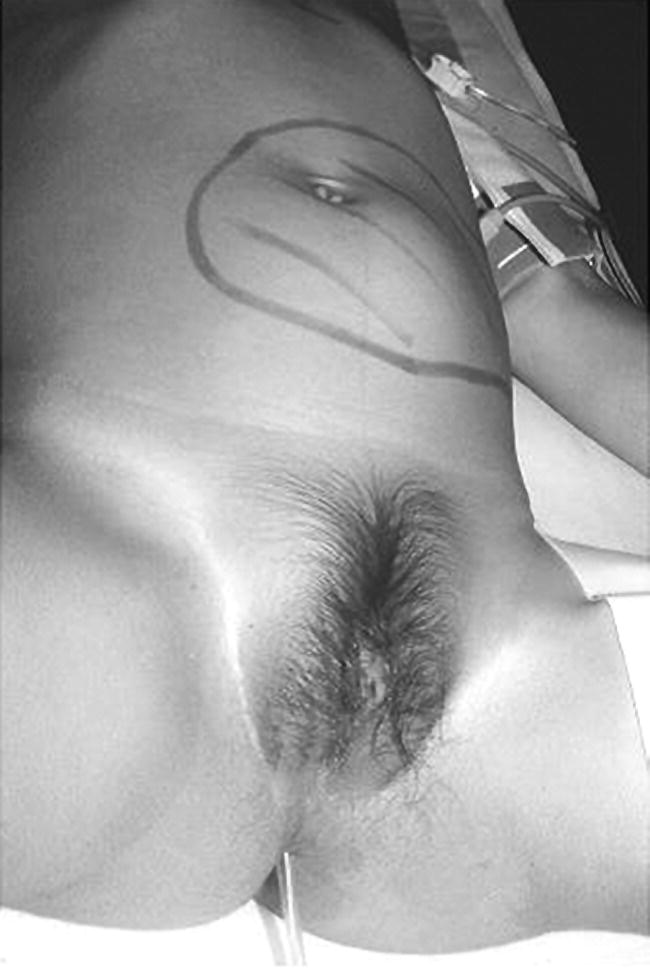
Investigations
Treatment and monitoring
Follow‐up
Diagnosis and management of sexual precocity in boys
Androgen‐mediated sexual precocity
True Central Precocious Puberty
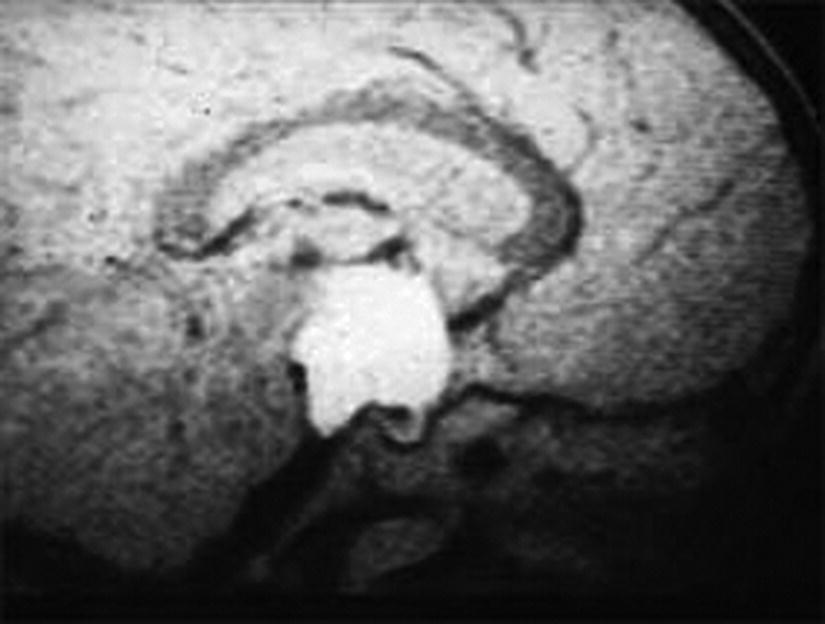
Diagnosis
Investigations
Treatment
Monitoring
Precocious Pseudopuberty
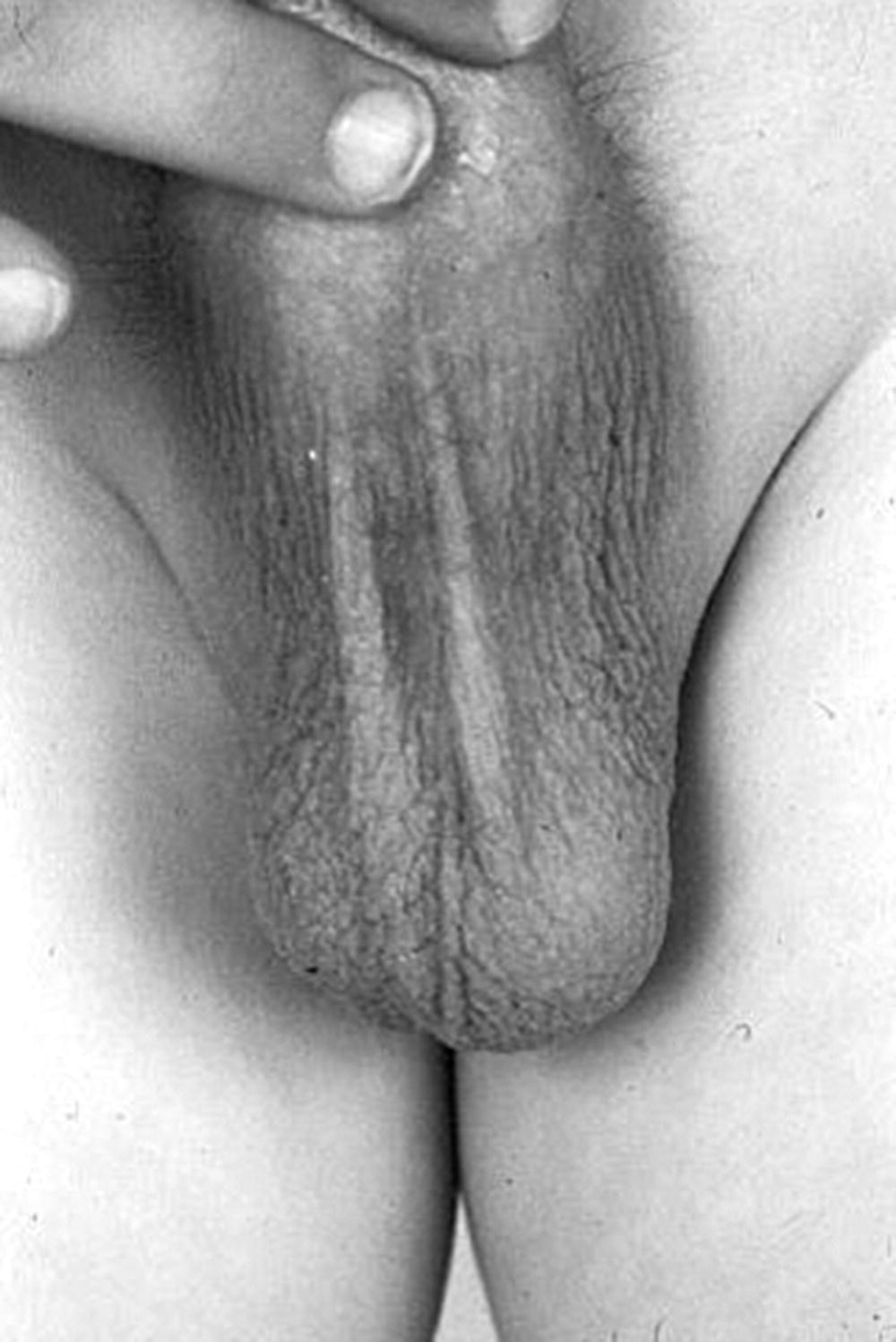
Investigations
Treatment
Adrenarche
Oestrogen‐mediated sexual precocity
Delayed puberty and pubertal failure
Definitions and classification
Central (both sexes)
Intact H–P axis
CDGA
Chronic systemic disease
Poor nutrition (including anorexia nervosa)
Psychosocial deprivation
Steroid therapy
Hypothyroidism
Impaired H–P axis
Tumours adjacent to H–P axis
Congenital anomalies
Irradiation
Trauma
GnRH/LH/FSH deficiency
Peripheral
Boys
Girls
Bilateral testicular damage
Disorders of sex development
Irradiation/chemotherapy
Syndromes associated with cryptorchidism
Polycystic ovary syndrome
Disorders of sex development (DSD)
Toxic damage
Galactosaemia
Other metabolic diseases (notably mitochondrial)
Irradiation/chemotherapy
Clinical assessment of boys and girls with delayed puberty
History
Examination
Investigations
Causes of delayed or absent puberty and pubertal failure
Central delay with intact axis
Constitutional Delay in Growth and Adolescence in Boys
Diagnosis
Investigation
Management
Follow‐up
Constitutional Delay in Growth and Adolescence in Girls
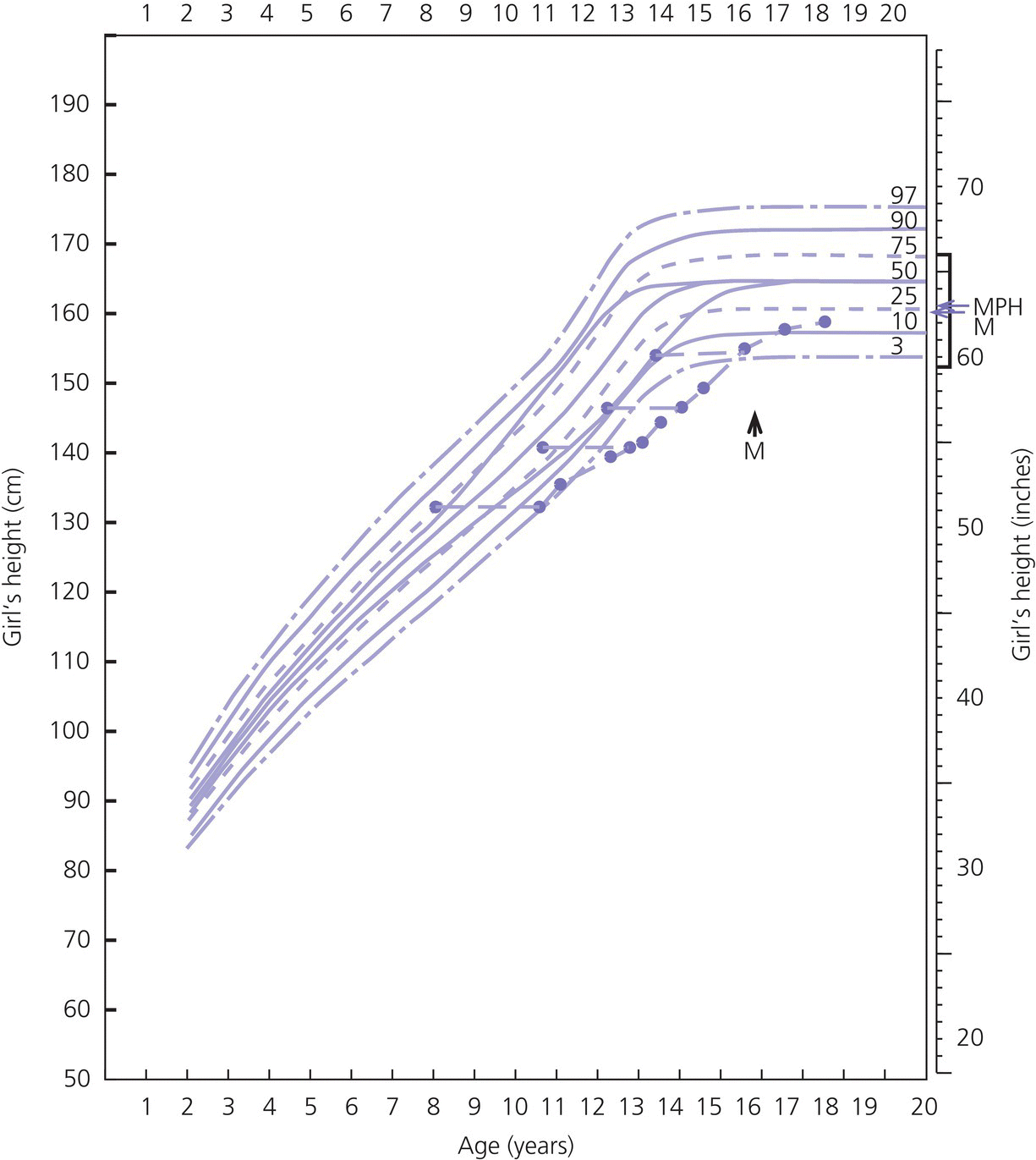
Diagnosis
Treatment
Delayed puberty caused by chronic disease
Impairment of the hypothalamo–pituitary–gonadal axis in boys
Central
Management
Peripheral
Investigations
Management
Treatment of Hypogonadism
Pubertal induction
Maintenance treatment
Impairment of the hypothalamo–pituitary–gonadal axis in girls
Central
Peripheral
Investigation
Management
Treatment
Monday to Thursday
Friday to Sunday
Duration (mo)
¼ patch:
No patch
6
¼ patch
¼ patch
6
½ patch
¼ patch
6
½ patch
½ patch
6
1 patch
1 patch
6
Dose
Tablets
Frequency
Equivalent daily dose
Duration (mo)
0.5 mg
½
Alternate days
0.25 mg
12
0.5 mg
½
Daily
0.5 mg
6
0.5 mg/1 mg
½, 1
Alternate days
0.75 mg
6
1 mg
1
Daily
1 mg
6
Other pubertal disorders
Prolactinoma
Menstrual problems
Investigation
Hirsutism and hypertrichosis
Diagnosis and Management
Investigations
Treatment
Polycystic ovary syndrome
Diagnosis and management
Treatment
Breast problems
Boys with Gynaecomastia
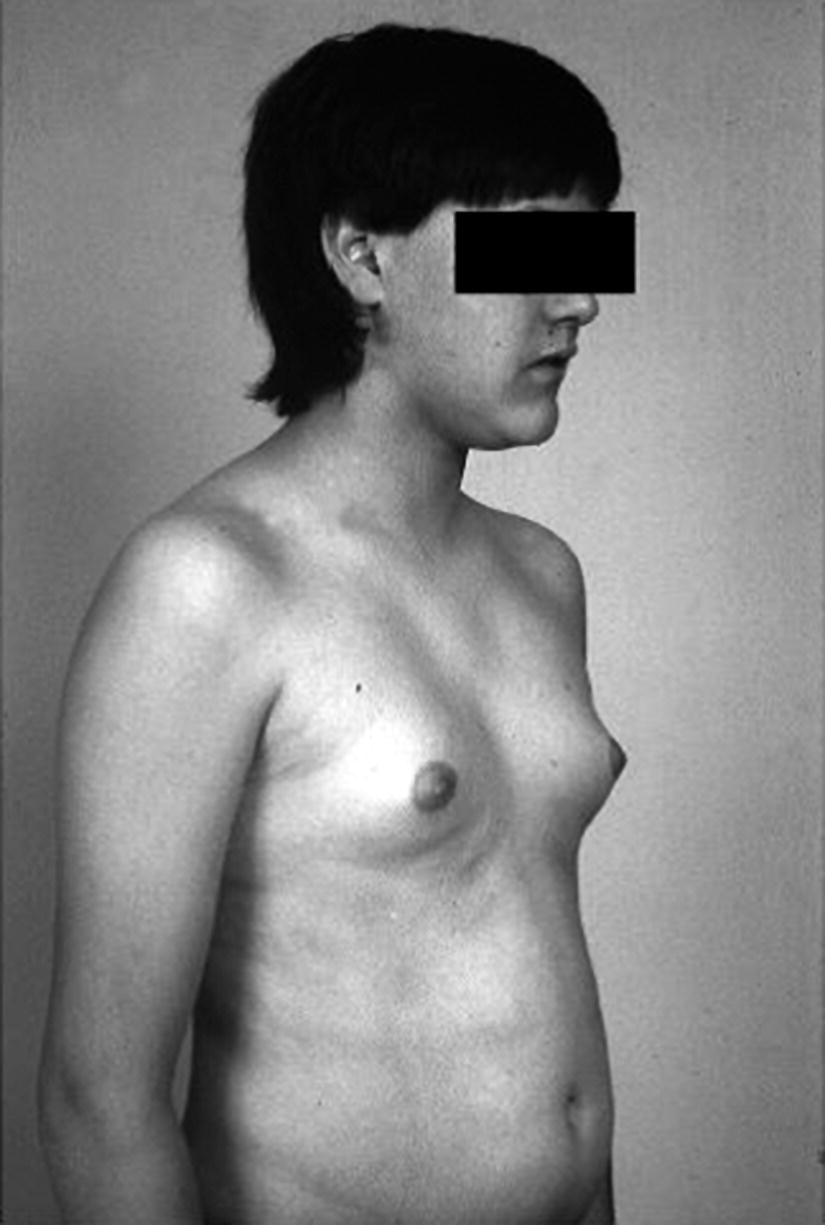
Treatment
Girls with Asymmetrical or Symmetrical Smallness or Largeness of Breasts
Symmetrical enlargement
Symmetrical smallness
Asymmetrical largeness or smallness
Future developments
Potential pitfalls
Controversial points
When to involve a specialist centre
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


