Prostate Cancer
Yu Chen
Vivek K. Arora
Charles L. Sawyers
Prostate cancer is the most common malignancy and the second leading cause of cancer death in Western men. It is highly heterogeneous with a disease specific mortality of one in seven. Many men survive decades without treatment, while others succumb quickly despite aggressive management. Among men who require systemic treatment, response rates to therapies are also highly variable—most notably some patients respond to androgen deprivation therapy for decades while a minority do not respond at all. Currently, the combination of clinical stage (tumor size on palpation), serum level of the prostate specific antigen (PSA), and histological grade (reported as Gleason score) is used to guide treatment decisions. Although useful for some clinical decisions, the modest predictive value of these parameters results both in overtreatment of many man and ineffective treatment for others. To achieve a “personalized medicine” approach to prostate cancer, it is important to define the repertoire of molecular lesions in prostate cancer, to identify the effect of these lesions on disease aggressiveness, and to identify therapies that have specific effectiveness against individual molecular lesions. Precedent for the ultimate success of this approach comes from other malignancies, such as the target specific kinase inhibitors erlotinib and PF-02341066 in lung cancers harboring epidermal growth factor receptor (EGFR) mutations and EML4-ALK fusions, respectively, the anti-HER2 antibody trastuzumab in breast cancers harboring HER2 amplification, and the kinase inhibitor PLX4032 in melanomas harboring BRAF mutations.1,2,3,4,5 In prostate cancer, recent work is beginning to define disease subtypes driven by different oncogenic genetic lesions. Ongoing research is expected to result in a more personalized treatment approach in prostate cancer, including who to screen, who to treat, and what form of treatment to use.
GENETIC PREDISPOSITION
Prostate cancer has a significant genetic component. A large Nordic study of 44,788 pairs of twins concluded that 42% of prostate cancers are attributable to genetic risk.6 Men with one, two, and three first degree relatives afflicted with prostate cancer have a 2-, 5-, and 11-fold risk of developing the disease, respectively.7 Given that large-scale screening of the general population is associated with small overall survival benefit and excess morbidity from curative surgery or radiation therapy for low risk disease,8,9 identification of genetic predispositions—especially of aggressive disease—could guide screening decisions.
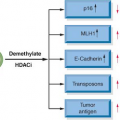
Early work identified rare mutations of several genes—ELAC2, MSR1, RNAseL—that confer increased risk of prostate cancer development but affect a small group of patients. Polymorphisms of genes in the androgen signaling pathway have been extensively studied with conflicting results.10 With the technology to detect single nucleotide polymorphisms (SNP), genome-wide association studies (GWAS) of large cohorts have discovered novel genomic regions associated with prostate cancer risk. A number of independent reports showed that polymorphisms of multiple loci on chromosome 8q24 account for 10% and 30% of prostate cancer cases among European and African ancestry, respectively.11,12,13,14,15 One challenge is to identify mechanisms by which these SNPs confer prostate cancer susceptibility since the 8q24 loci are located at a “gene desert.” Recent work suggests that at least one loci may serve as an enhancer for MYC, which is approximately 300 kb away.16 In addition, other loci have been recently discovered,17,18 but none have shown a significant enough increase in the odds ratio of developing cancer to warrant screening of the general population. There is some evidence that simultaneous presence of multiple risk-associated SNPs in the same individual is associated with a significantly higher risk of prostate cancer.19
GENETIC LANDSCAPE OF PROSTATE CANCER
Like other epithelial tumors, many prostate cancers have a large number of genetic and epigenetic lesions associated with hallmark properties of
malignancies.20 Types of lesions include overexpression of oncogenes and underexpression of tumor suppressor genes, genomic amplifications and deletions, translocations, and, more rarely, point mutations. One recent large-scale study that combines gene expression profiling, comparative genomic hybridization (array CGH) to characterize amplifications and deletions, and targeted resequencing for somatic mutations has shed light onto the genetic landscape of prostate cancer.21 On average, prostate cancer is characterized by a relatively low mutation rate of genes commonly mutated in other malignancies, such as KRAS, BRAF, or p53, but a relatively large number of genomic alterations (Fig. 22.1). Recurrent amplifications and deletions, some focal, covering single to a handful of genes, and others spanning entire arms covering thousands of genes, are present in many tumors at diagnosis. On average, higher grade and stage tumors contain a greater number of genomic alterations. The most common genomic alteration is loss in the chromosomal arm 8p followed by a gain of chromosomal 8q. Some focal genomic alterations, such as deletion of PTEN or deletion of the interstitial region between TMPRSS2 and ERG that creates the TMPRSS2-ERG fusion gene, results in a clearly established oncogenic driver lesion. But for most genomic alterations, the specific genes responsible for oncogenesis are a subject of speculation.
malignancies.20 Types of lesions include overexpression of oncogenes and underexpression of tumor suppressor genes, genomic amplifications and deletions, translocations, and, more rarely, point mutations. One recent large-scale study that combines gene expression profiling, comparative genomic hybridization (array CGH) to characterize amplifications and deletions, and targeted resequencing for somatic mutations has shed light onto the genetic landscape of prostate cancer.21 On average, prostate cancer is characterized by a relatively low mutation rate of genes commonly mutated in other malignancies, such as KRAS, BRAF, or p53, but a relatively large number of genomic alterations (Fig. 22.1). Recurrent amplifications and deletions, some focal, covering single to a handful of genes, and others spanning entire arms covering thousands of genes, are present in many tumors at diagnosis. On average, higher grade and stage tumors contain a greater number of genomic alterations. The most common genomic alteration is loss in the chromosomal arm 8p followed by a gain of chromosomal 8q. Some focal genomic alterations, such as deletion of PTEN or deletion of the interstitial region between TMPRSS2 and ERG that creates the TMPRSS2-ERG fusion gene, results in a clearly established oncogenic driver lesion. But for most genomic alterations, the specific genes responsible for oncogenesis are a subject of speculation.
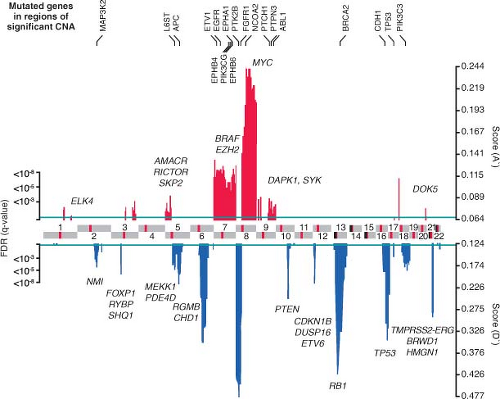 FIGURE 22.1 Genomic aberrations in prostate cancer. Regions of amplification (red) or deletion (blue) with false discovery rate (FDR) 10% or less are plotted, with chromosomes indicated at the center and centromeres in red. Genes with somatic nonsynonymous mutations are listed on top (black). Additional genes of interest targeted by copy-number alterations alone are also indicated (gray) (From ref. 21 with permission). |
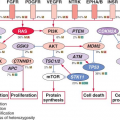
Although many individual genes are rarely affected in prostate cancer, several key oncogenic and tumor suppressor pathways are commonly involved when the individual genes in each pathway are considered collectively. Four pathways—the phosphatidylinositol 3-phosphate kinase (PI3K)/AKT pathway, the RAS/MAP kinase pathway, the retinoblastoma pathway, and the androgen receptor (AR) pathway—are altered in one-third of primary cancers and almost all of metastatic lesions. This suggests that in prostate cancer, different individual genes in the pathway may be targeted to activate or suppress a common pathway lesion (Fig. 22.2). The sections below focus on those pathways where prostate cancer is most validated.
Androgen Receptor Pathway
It has been 70 years since Higgins and Hodges made the seminal observation that prostate cancers
regress after surgical orchiectomy or suppression of androgen production by administration of exogenous estrogens.22 These observations proved that prostate cancer is an androgen-dependent malignancy, and androgen deprivation therapy became the first targeted therapy in oncology—leading to a Nobel Prize for Charles Huggins in 1966.
regress after surgical orchiectomy or suppression of androgen production by administration of exogenous estrogens.22 These observations proved that prostate cancer is an androgen-dependent malignancy, and androgen deprivation therapy became the first targeted therapy in oncology—leading to a Nobel Prize for Charles Huggins in 1966.
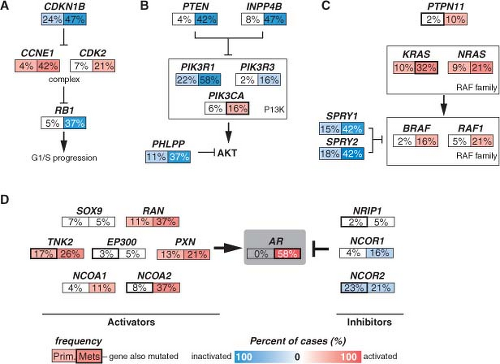 FIGURE 22.2 Alterations in the retinoblastoma (A), phosphatidylinositol 3-kinase (PI3K) (B), RAS/MAP kinase (C), and androgen receptor (AR) (D) pathways in prostate cancer. Alteration frequencies are shown for individual genes and for the entire pathway in primary and metastatic tumors. Alterations are defined as those having significant up- or down-regulation compared with normal prostate samples, or by somatic mutations, and are interpreted as activation (red) or inactivation (blue) of protein function. (From ref. 21 with permission.) |
Androgens exert their cellular actions through the AR, a 110 kb steroid receptor transcription factor, located on Xq12.23,24 Upon binding androgen, AR mediates transcription of a number of genes involved in survival and differentiation of prostate epithelial cells. AR activity is required for development of both normal and malignant prostate tissue as men with germline AR inactivating mutations or men orchiectomized at a young age do not develop a prostate gland. AR plays a critical role in early pathogenesis as well as progression to advanced disease, and drugs that inhibit AR function remain the primary treatment for advanced prostate cancer.
Early Pathogenesis
Several lines of evidence suggest that enhanced AR signaling may play a causative role in prostate cancer initiation. Polymorphisms of three genes in the AR pathway, AR itself, CYP17, and SRD5A2, are low penetrant risk factors for prostate cancer development in some but not all studies.6 Overexpression of AR in both mouse and human primary prostate cells confers tumor forming ability when injected orthotopically into mice.25,26 In rat models of carcinogen-induced prostate cancer, implantation of testosterone pellets results in an increased number of tumors.27 Transgenic mice that overexpress AR in the prostate develop prostatic intraepithelial neoplasia (PIN).28
Pathway analysis from a recent large-scale prostate cancer genomic project found AR pathway alterations in 60% of primary tumors. The most commonly altered AR pathway member was NCOA2 (also called SRC2), an AR coactivator that potentiates AR transcriptional output. Copy gains or somatic mutations of the NCOA2 gene were found in approximately 20% of primary tumors. In addition, several other AR coactivators showed increased expression, while AR corepressors were down-regulated (Fig. 22.2D).
Cancer Progression and Castration-Resistant Growth
Because of the dependence of the prostate lineage on AR function, chemical castration through
suppression of testicular function has become the mainstay in systemic treatment of prostate cancer. In normal prostate tissue, castration results in atrophy and cessation of PSA production indefinitely until restoration of testosterone levels. However, prostate cancers progress to the lethal castration-resistant prostate cancer (CRPC) after a variable duration of response. Cancer progression is usually accompanied by restoration of PSA secretion, indicating reactivation of AR activity. Thus, instead of bypassing the requirement for AR function, most castrate-resistant tumors have reactivation of the AR pathway, likely through multiple mechanisms.23
suppression of testicular function has become the mainstay in systemic treatment of prostate cancer. In normal prostate tissue, castration results in atrophy and cessation of PSA production indefinitely until restoration of testosterone levels. However, prostate cancers progress to the lethal castration-resistant prostate cancer (CRPC) after a variable duration of response. Cancer progression is usually accompanied by restoration of PSA secretion, indicating reactivation of AR activity. Thus, instead of bypassing the requirement for AR function, most castrate-resistant tumors have reactivation of the AR pathway, likely through multiple mechanisms.23
Alterations of AR itself represent the best characterized mechanism of castration resistance. Mutations of AR are detected in up to 10% of CRPCs and result in activation by other hormone ligands such as corticosteroids or, paradoxically, by antiandrogens.29 Overexpression of AR is seen in the majority of CRPCs, and the AR gene is amplified in approximately 30% of cases.30,31,32 AR overexpression is sufficient to activate AR in the milieu of castrate levels of androgens and to convert the antiandrogen, bicalutamide, into a weak agonist.33 Alternative splicing of AR mRNA can lead to a truncated protein containing the N-terminal transcriptional activation and DNA binding domains but missing the ligand binding domain. Some of these variants have constitutive transcriptional activity in the absence of androgens and can be detected in CRPC samples. Overexpression of at least two variants can confer castration resistance in preclinical models.34,35,36
Alterations in AR pathway genes are found in 100% of metastases and may enhance AR activity in the presence of castrate androgen levels. NCOA2, for example, increases the magnitude of AR signaling across a broad range of androgen concentrations. Several other androgen receptor coactivators, including NCOA1, TNK2, and EP300, are up-regulated in metastatic disease, while AR corepressors, including NRIP1, NCOR1, and NCOR2, are down-regulated (Fig. 22.2D).
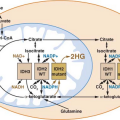
Intratumoral androgens are not completely eliminated by castration and may be increased in CRPC.37 Direct measurements of intratumoral androgens by mass spectrometry showed that castration-resistant metastatic tumors in men treated with gonadotropin-releasing hormone (GnRH) agonists such as leuprolide have higher levels of testosterone than primary tumors in untreated men.38 There is some evidence that CRPC may synthesize androgens to activate AR in an autocrine loop. Two expression profiling studies that compared metastatic CRPC with primary tumors have shown that enzymes involved in androgen synthesis are up-regulated in CRPC. Holzbeierlein et al.32 found overexpression of enzymes involved in synthesis of cholesterol, the common steroid precursor, from acetyl-CoA, and Stanbrough et al.39 found overexpression of enzymes involved in synthesis of testosterone and the more potent androgen dihydrotestosterone (DHT) from cholesterol. A third study of patient samples at all stages of disease found an abundance of the enzymes AKR1C3 and SRD5A1 necessary for conversion of androstenedione to DHT. However, the samples lacked high expression of enzymes necessary for de novo steroidogenesis.40 These data suggest that autocrine androgen synthesis may allow tumors to grow despite low serum androgen levels, but that this process may in part be dependent on adrenal precursors (Fig. 22.3).
Multiple kinase signaling pathways have been implicated in CRPC—most notably the HER2 and AKT pathways—with evidence that they exert their effects, in part, through modulation of AR activity. The HER2 receptor tyrosine kinase is progressively overexpressed in more advanced CRPC, though it is seldom amplified as seen in breast cancer. In experimental systems, forced overexpression of HER2 results in castration resistance, while pharmacologic inhibition or protein knockdown results in growth suppression.41 Loss of PTEN and activation of the AKT pathway, commonly seen in CRPC (Fig. 22.2B), can contribute to castration resistance. Primary tumors with loss of PTEN have lower response rates to neoadjuvant bicalutamide.42 Prostate cancer in mice with genetically engineered loss of PTEN rapidly develop castration resistance, and PTEN knockdown in murine hormone-sensitive prostate cancer cells confers castration-resistant growth.43,44
Androgen Receptor Targeting
Androgen deprivation therapy (ADT), most commonly delivered by administration of GnRH such as leuprolide, remains the cornerstone of prostate cancer systemic therapy. With the discovery that CRPC is associated with reactivation of AR and remains AR dependent, there have been many recent efforts to develop alternative AR targeting strategies (Fig. 22.3).
Antiandrogens
Antiandrogens compete with endogenous androgens for the ligand binding pocket of AR. Similar to androgens, antiandrogens promote translocation of AR from the cytoplasm into the nucleus and DNA binding but induce conformational changes that prevent optimal transcriptional activity. In the United States, there are three nonsteroidal antiandrogens currently in use—flutamide, bicalutamide, and nilutamide. Although generally active upon treatment initiation, each compound has been associated with the antiandrogen withdrawal response, whereby treated patients with progressive disease derive clinical benefit when the antiandrogen is stopped,45 indicating that they
have potential to act as AR agonists despite their intended role as antagonists. Indeed, overexpression of AR can convert bicalutamide from an antagonist into an agonist.33
have potential to act as AR agonists despite their intended role as antagonists. Indeed, overexpression of AR can convert bicalutamide from an antagonist into an agonist.33
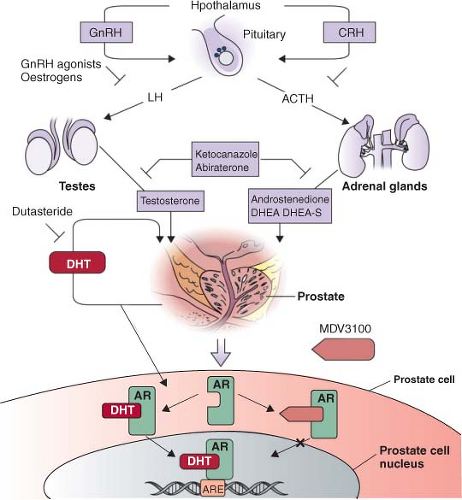 FIGURE 22.3
Get Clinical Tree app for offline access
Related posts:Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree


|