Chapter 8 Principles of Chemotherapy
Although the earliest clinical manifestation of cancer is a mass or tumor, in many patients cancer is often not a localized disease at the time of its discovery. In most patients, the disease has already spread to lymph nodes and other distant sites, even though clinical imaging and random biopsies may fail to disclose its presence. For more than a century, thoughtful investigators have sought systemic treatments for cancer, often using as their paradigms the lessons of infectious illnesses and nutritional disease. Vaccines, natural extracts, vitamins, and synthetic chemicals were touted as cures. However, not until the initial trials of alkylating agents by scientists and clinicians at Yale University, first described in 1946, was there proof that a systemically administered agent could cause regression of a human tumor, which in this case was a mediastinal mass in a patient with Hodgkin’s disease.1 The clinical studies of nitrogen mustard, based on observations of the toxicity of mustard gases in World War I, lacked a clear conceptual basis. Although it was known at the time that these compounds were highly reactive with proteins, nucleic acids, and other electron-rich molecules, the specific intracellular target was not identified until more than a decade later, and we are only now beginning to understand the reasons for the selective action of alkylating agents on tumor cells.
This early experience with alkylating agents led a number of bold investigators to undertake an alternative approach, the synthesis of compounds that would act as fraudulent counterparts of natural metabolites known to stimulate cancer cell growth. Among these metabolic targets, vitamins and nucleic acid bases proved to be most vulnerable (Fig. 8-1). In the late 1940s, scientists from American Cyanamid synthesized analogs of folic acid, a vitamin known to stimulate the proliferation of cancer cells in culture and in humans. Sidney Farber tested these analogs and found striking but short-lived responses to aminopterin and to the closely related compound methotrexate in children with acute lymphoblastic leukemia (ALL).2 Prospects for the treatment of cancer with drugs dramatically escalated in the following decade. Hitchings, Elion, and others at Burroughs Wellcome experimented with a series of purine analogs and found antitumor effects in animals, leading to the successful development of 6-thioguanine and 6-mercaptopurine. Soon afterward, corticosteroids were discovered as anti-inflammatory agents and were found to kill malignant lymphocytes in children with leukemia and lymphoma.3
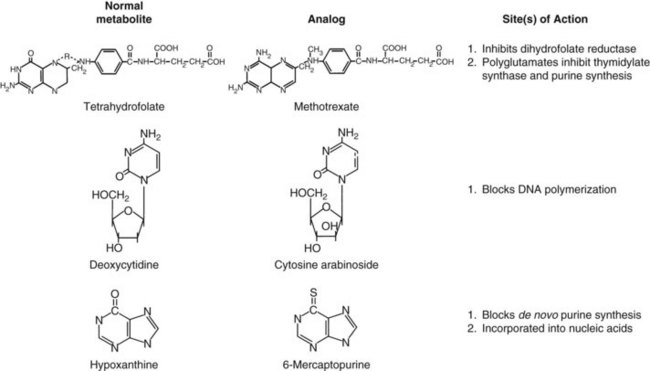
Equally rewarding efforts in the field of natural product chemistry yielded additional new anticancer drugs with unique mechanisms of action. In the course of their search for natural products for the treatment of diabetes, scientists at Eli Lilly discovered the antimitotic and antitumor properties of the vinca alkaloids.4 Shortly thereafter, analysis of fermentation broths led to the discovery of mitomycin C, a novel alkylating agent5; bleomycin, a DNA-cleaving peptide6; and the anthracyclines, which inhibit topoisomerase II,7 an enzyme that relaxes DNA supercoils during transcription and replication. In the 1960s, further efforts in plant chemistry led to the isolation of paclitaxel,8 a drug that inhibits formation of the mitotic spindle, and the camptothecins, which block the action of topoisomerase I.9 However, paclitaxel and the camptothecins did not reach the clinic until 10 to 20 years later.
Models for Chemotherapy
In parallel with the work that led to cytotoxic chemotherapy, Skipper and Perry10 at the Southern Research Institute characterized murine tumor models, notably the L1210 and P388 leukemias, as well as murine solid tumors such as sarcoma 180. Their model systems allowed reproducible, quantitative chemotherapy experiments to be performed in mice.10 They tested various strategies for treatment and established a rational basis for understanding the kinetics of cell kill, the evaluation of drug combinations, and the mechanisms of drug resistance. Important concepts of cancer chemotherapy came from their experiments, which established the theoretical basis for rational combination chemotherapy and the cure of ALL in children. Skipper’s experiments with murine leukemias established the following principles:
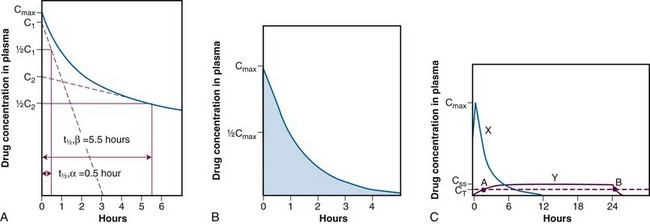
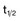 ,α) is the time for C1 to decay to
,α) is the time for C1 to decay to 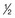 C1. The terminal (β) phase half-life (
C1. The terminal (β) phase half-life (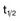 ,β) is the time for C2 to decay to
,β) is the time for C2 to decay to 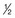 C2. This biphasic behavior results from distribution of the drug among rapidly and slowly perfused regions of the body, as well as its elimination. B, Drug concentration in plasma versus time is plotted on linear axes. The blue shaded area is the area under the curve (AUC); it represents the integral of drug concentration over time. The AUC is a measure of total systemic exposure to the drug. C, Linear plots of drug concentration versus time are illustrated for a rapid intravenous injection (X) and a 24-hour continuous infusion (Y) of the same total dose of drug (the AUCs are equivalent). Notice that the duration of drug concentrations above the threshold for cytotoxicity (CT) is much longer with the continuous infusion (represented as B-A) than with the bolus administration. Conversely, the maximum plasma concentration achieved by bolus administration (Cmax) is much larger than that of the continuous infusion (CSS).
C2. This biphasic behavior results from distribution of the drug among rapidly and slowly perfused regions of the body, as well as its elimination. B, Drug concentration in plasma versus time is plotted on linear axes. The blue shaded area is the area under the curve (AUC); it represents the integral of drug concentration over time. The AUC is a measure of total systemic exposure to the drug. C, Linear plots of drug concentration versus time are illustrated for a rapid intravenous injection (X) and a 24-hour continuous infusion (Y) of the same total dose of drug (the AUCs are equivalent). Notice that the duration of drug concentrations above the threshold for cytotoxicity (CT) is much longer with the continuous infusion (represented as B-A) than with the bolus administration. Conversely, the maximum plasma concentration achieved by bolus administration (Cmax) is much larger than that of the continuous infusion (CSS).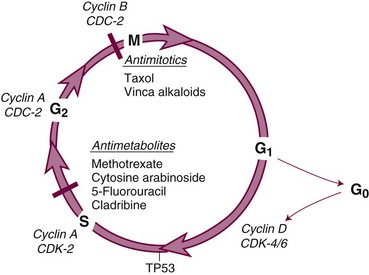
These principles profoundly influenced all aspects of clinical chemotherapy, including regimen design, the use of drugs in combination, adjuvant chemotherapy, and high-dose chemotherapy. Relying on these principles, the cure of ALL was accomplished through the efforts of Frei and Freireich at the National Cancer Institute and Holland at Roswell Park, who developed effective combination therapy; Pinkel and colleagues at St. Jude Hospital in Memphis, who identified the central nervous system as a sanctuary for leukemic cells; and the national pediatric cooperative groups, which tested these insights in randomized clinical trials.11 These investigators combined a knowledge of the drugs and the disease to establish principles for leukemia therapy that persist to this day: intensive, marrow-ablative induction therapy, followed by consolidation to further reduce the leukemic cell population; central nervous system prophylaxis with irradiation and methotrexate; and maintenance chemotherapy to eliminate the last logs of tumor cells. In the course of their work, they identified the importance of supportive care measures, including aggressive broad-spectrum antibiotic treatment of febrile episodes in neutropenic patients, platelet support to prevent bleeding due to thrombocytopenia, and management of opportunistic infection. Each of these insights, further refined by the use of new antibiotics and bone marrow colony-stimulating factors, has become a basic component of modern chemotherapy.
Solid Tumor Chemotherapy
The principles enumerated earlier have been applied with greatest success to the treatment of aggressive and rapidly proliferating tumors such as leukemias, lymphomas, and testicular tumors. The development of drug therapy for the more common solid tumors has taken a slower and more tortuous course. Drugs identified in mouse leukemia screening systems have been less effective against most solid tumors; notable exceptions are choriocarcinoma, testicular cancer, and the lymphomas, all of which can be cured with cycles of intensive chemotherapy. However, chemotherapy has had only limited impact on the common solid tumors in humans, particularly in widely metastatic, end-stage disease. Rational synthesis of 5-fluorouracil (5-FU) by Heidelberger and associates12 led to the first modestly effective antimetabolite for colon and breast cancers. Other important clinically effective drugs were identified through random screening efforts and serendipity: doxorubicin and cisplatin in the early 1970s, etoposide and paclitaxel in the late 1980s, and the camptothecin analogs and gemcitabine in the 1990s.13
An important conceptual breakthrough in solid tumor chemotherapy was the proposal to employ drugs in the adjuvant setting after removal of the primary tumor in patients at high risk for relapse. The work of Fisher,14 Bonnadonna, and others established that drugs capable of producing partial responses in advanced disease could cure one third of women with stage II breast cancer, who were otherwise destined to suffer distant recurrence and death. The conceptual basis for this strategy is the increased probability of curing metastatic disease when the tumor burden is small and tumor cells are actively proliferating and are, therefore, more susceptible to cytotoxic drugs. Drugs have a firmly established role in the treatment of lung, breast, and colorectal cancers after or before surgery.
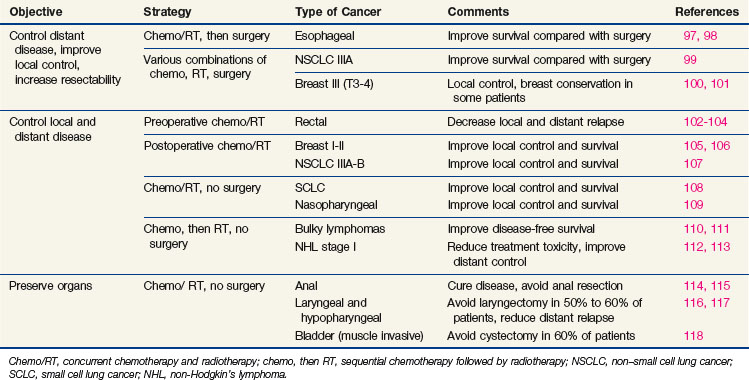
Newer concepts aimed at improving local control of otherwise inoperable tumors have led to so-called neoadjuvant, or induction, therapy.15 In this strategy, drugs are used alone or in combination with radiation therapy, before surgery, in the initial treatment of locally advanced tumors of the breast, head and neck, bladder, rectum, and lung (Table 8-1). This preoperative therapy reduces the size of otherwise unresectable tumors to a point where surgical removal is feasible or, in some cases, unnecessary.
Because drugs have become an integral part of the initial therapy of many patients with cancer, it is essential that the medical oncologist, surgeon, and radiation oncologist understand the principles of chemotherapy and the specific features of the commonly used agents. This chapter explains the conceptual basis for the use of cancer chemotherapeutic drugs and provides detailed information for understanding their actions, toxicities, and late side effects.16
The Basis Of Cancer Chemotherapy: Cancer Cell Biology
Every phase of chemotherapeutic research, from discovery to clinical application, is based on the concepts of cancer cell biology, and drug research has evolved as knowledge of biology has progressed. The essential properties of the cancer cell, properties that distinguish the cell from its normal counterpart and that form the basis for treatment, are excess proliferation, defective repair of DNA leading to high mutation rates and population diversity, reduced rates of apoptosis (i.e., programmed cell death), invasive capacity, ability to induce nutrient vessels, ability to escape immune surveillance, and ability to metastasize. Each of these properties has been the starting point for drug discovery efforts,17 as illustrated in Table 8-2. Most cancer cells display defects in DNA repair that generate a diversity of subclones, increase their adaptation to diverse and at times adverse environments, and increase the probability of drug resistance. However, with a few notable exceptions, the successful cytotoxic drugs have attacked only the first of these properties, proliferation. The new generation of targeted agents is expanding the horizon for cancer treatment by addressing the full circle of biologic changes in human tumors.
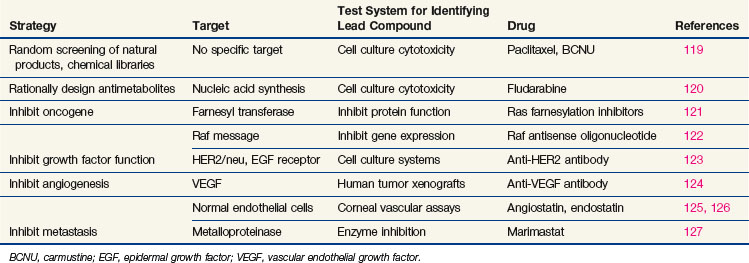
Drugs That Target Dna Synthesis And Mitosis
Although much of the effort in modern cancer drug development is directed at targets that regulate growth signal transmission, most of the successful drugs in current clinical use act directly on the synthesis or integrity of DNA. These drugs inhibit synthesis of DNA or its precursors, block cell division, inhibit necessary changes in DNA topology, or covalently bind to DNA, causing strand breaks or miscoding. All such drugs affect the integrity of DNA, and in the presence of the normal machinery for monitoring DNA integrity, they induce apoptosis. Unfortunately, they also tend to be cytotoxic toward normal cells. The reasons for selectively greater effects of cytotoxic drugs on malignant versus normal cells, as is apparent in the cure of certain malignant tumors, are poorly understood, although the process of malignant transformation may enhance sensitivity to DNA damage by virtue of defects in DNA repair.18 For example, many of the defective genes responsible for inherited cancer syndromes, such as BRCA1 and BRCA2 genes, mismatch repair genes, TP53, and nucleotide excision repair, create sensitivity to agents that inhibit various aspects of DNA repair. Another key process in malignant transformation is the loss of cell cycle controls that allow DNA repair and prevent apoptosis.
The cell cycle and its primary controls are shown in Figure 8-3. Antimetabolites such as methotrexate, 5-FU, cytosine arabinoside, gemcitabine, and the purine antagonists require cells to be actively proliferating to kill them; therefore, they are cell cycle dependent. Many of these agents, including cytosine arabinoside, fludarabine phosphate, and cladribine, must be incorporated into DNA to be cytotoxic. Alternately, agents such as etoposide and doxorubicin produce irreparable DNA strand breaks at any stage of the cell cycle, although these breaks become lethal only as the cell enters DNA synthesis. Still others, such as alkylating agents and platinum compounds, bind covalently to DNA and produce strand cross-links and strand breaks. Their toxicity seems less dependent on the stage of the cell cycle; some agents of this class, such as the nitrosoureas, are equally capable of killing nondividing and dividing cells. Antimitotic agents, constituting a separate class of drugs, block the formation or dissociation of the mitotic spindle through their effects on microtubules and thereby prevent separation of chromosomes to the daughter cells. Drugs of this class are therefore most effective against cells that enter the mitotic phase of the cell cycle.
Once it has been damaged through its encounter with a cytotoxic drug, the cancer cell has several options, and its eventual viability depends on which pathway it takes. If the normal monitors for genomic integrity (including most prominently the product of the TP53 gene) are intact, the cell may halt further progression in the cell cycle while its DNA is repaired. If the damage is sufficiently severe, intact TP53 may initiate apoptosis. If, however, the TP53 function is absent, cell cycle progression may continue despite drug-induced DNA damage, and the cell may prove viable. In most experimental settings, lack of wild-type TP53 is associated with drug and radiation resistance, but with certain drugs (e.g., paclitaxel), loss of the G1-S checkpoint function does not interfere with response.19 Paradoxically, certain DNA repair defects, such as those commonly found in mismatch repair (colon cancer) or excision repair (ovarian cancer), may be associated with resistance to platinum analogs, antimetabolites, and alkylating agents, perhaps by preventing recognition of DNA adducts and failing to initiate apoptosis.
Drugs That Target the Controls of Cell Proliferation and Cell Death
In the past few years, drug discovery efforts have targeted specific pathways and mutations that promote proliferation (see Table 8-2). Among these mutations, a number of pharmaceutical companies have focused attention on dominant mutations, such as the activation of RAS in pancreatic, colon, and bladder cancer or the activation of cell surface growth factor pathways such as the epidermal growth factor receptor (EGFR) and other tyrosine kinase receptors in epithelial tumors. Their choice of targets (activating mutations or oncogenes) was largely driven by the difficulty of developing a product or strategy to replace a missing suppressor gene product such as TP53 or CDKN2A (previously designated p16).
Inhibiting the function of a dominant gene product is a classic problem in drug discovery. The first efforts at targeted inhibition of a signaling pathway led to development of inhibitors of the KRAS protein, a signal transducer that links growth factor receptors to downstream molecules. KRAS is mutated in 80% to 90% of pancreatic cancers and provides the proliferative drive in a number of human cancers.22 The RAS protein is activated by a series of enzymatic steps that attach a lipid tail, a farnesyl group, to its near-terminal cysteine; methylate the cysteine; and cleave off the three terminal amino acids (Fig. 8-4). In this case, drug design seemed to be a relatively straightforward task in which a key enzymatic process or receptor is identified and a potent and specific inhibitor is developed. Several companies focused on the farnesylation step in RAS processing as a target for inactivating RAS and have found that inhibitors of farnesylation cause regression of experimental tumors. However, these drugs failed in clinical trials because the inhibitors lacked specificity for farnesylation of RAS, and alternative pathways for lipid modification of the gene product exist in cells.
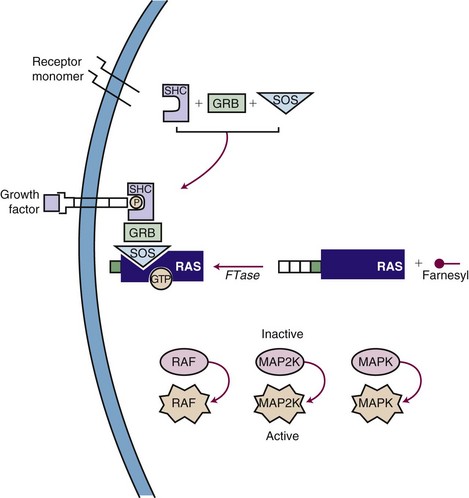
A second pathway of great interest in modern cancer drug development is the RB pathway, which provides a brake on cell proliferation. Mutations may affect any one of the several genes and their proteins (e.g., CDKN2A, CDK4, CCND [cyclin D], RB1) that regulate phosphorylation and inactivation of the RB protein and free tumor cells from controls. These mutations have been found in a variety of human tumors and seem to be an essential step in converting tumors to a highly malignant phenotype. Several pharmaceutical firms have identified candidate inhibitors of cyclin-dependent kinases (CDKs), including CDK4 and CDK9, although most such compounds lack exquisite specificity for their target. One such compound, flavopiridol, an inhibitor of CDK9, has shown promising activity in chronic lymphocytic leukemia, particularly in patients with high-risk features and refractory disease.23 The most promising approach to inhibiting regulatory pathways of cell proliferation has resulted from targeting receptor tyrosine kinases. These receptors are constitutively activated by mutations (EGFR in non–small cell lung cancer [NSCLC], BRAF in melanoma and other solid tumors), amplifications (HER2/neu in breast cancer), and activating translocations (EML-4-ALK in NSCLC, BCR-ABL in chronic myelogenous leukemia [CML]). The net effect is to create a cell under constant proliferative stimulus and addicted to the aberrant signal; it dies when the signal is interrupted. These examples have led to a search for additional targets, exploration of both small molecules and antibodies, and a search for therapeutic agents. Some of the agents, particularly the monoclonal antibodies, have proven synergistic with chemotherapy.
Epidermal growth factor receptor (EGFR) inhibitors constitute another class of agents that have proved useful in NSCLC. EGFR is overexpressed in many solid tumors. It plays a critical role in cell cycle progression, proliferation, and survival. Its effects are mediated through RAS. Activation of EGFR is also associated with production of vascular endothelial growth factor (VEGF), leading to angiogenesis and facilitating metastasis. Although the U.S. Food and Drug Administration (FDA) approved the small-molecule tyrosine kinase inhibitor gefitinib as a single agent for the third-line treatment of NSCLC in 2003, it later limited availability of gefitinib to patients continuing to derive benefit. This change occurred in response to new trials that failed to demonstrate a survival benefit in unselected patients with NSCLC24,25 (Table 8-3). Another oral EGFR tyrosine kinase inhibitor, erlotinib, demonstrated an improvement in overall and progression-free survival rates compared with placebo, in patients with NSCLC who had failed at least one prior chemotherapy.26 Surprisingly, however, when gefitinib or erlotinib was added to standard chemotherapy in the first-line management of patients with advanced NSCLC, there was no improvement in response or survival rates.27,28–30
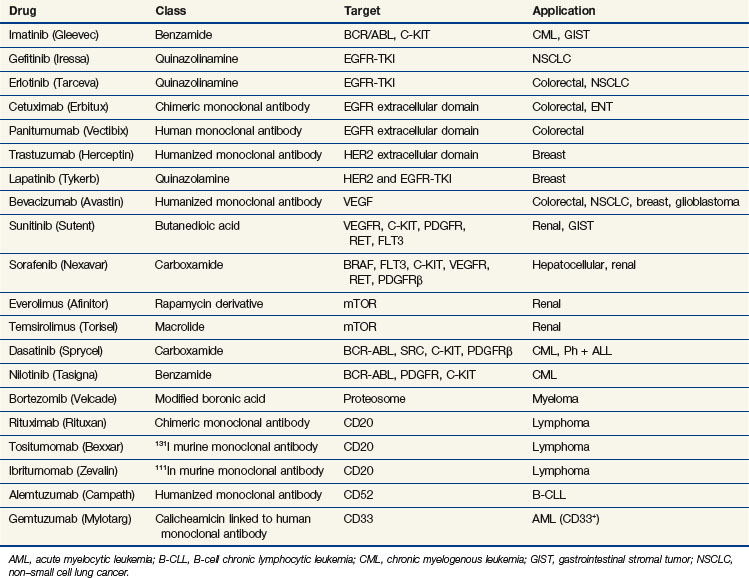
Subsequent trials have revealed that this new class of drugs is highly effective for the small subset of NSCLC patients with tumors that harbor activating mutations in EGFR. Although 40% to 80% of NSCLCs overexpress EGFR, only about 10% to 19% of patients with chemotherapy-refractory NSCLC respond to this therapy.31,32,33 Responses are more frequent in women, nonsmokers, and patients with bronchoalveolar carcinoma or adenocarcinoma with bronchoalveolar features.34 Mutations caused by in-frame deletions or amino acid substitutions clustered around the ATP-binding pocket of the tyrosine kinase domain of EGFR strongly predict the response to gefitinib, suggesting a gain of function and addiction to EGFR signaling and increased sensitivity to gefitinib.35,36 Patients with such mutations of EGFR have an approximately 70% chance of responding to erlotinib or gefitinib.37
In 2004, the FDA approved cetuximab, a chimeric IgG1 monoclonal antibody targeting the extracellular portion of the EGFR, for the treatment of irinotecan-refractory colorectal cancer. The addition of irinotecan to cetuximab in this population results in nearly doubled response rates, suggesting reversal of resistance.38 Panitumumab is another commercially available monoclonal antibody targeting the EGFR, which resulted in an improvement in progression-free survival rates compared with placebo alone in chemotherapy-refractory patients.39 Cetuximab alone results in an improvement in survival rates compared with placebo in chemotherapy-refractory patients.40 Several studies have demonstrated a superior response in patients who develop the typical acneiform rash associated with these agents. More importantly, several trials have proven that patients who have activating mutations of KRAS or BRAF derive no benefit from these antibodies, and, in fact, their inclusion may have a negative effect on response.41,42 In studies evaluating the combination of bevacizumab and chemotherapy, the addition of anti-EGFR antibodies resulted in a deleterious response, most strikingly in KRAS-mutant tumors.43,44 Cetuximab has also proved to be an active agent in the management of squamous cell carcinoma of the head and neck, with or without irradiation.45,46 Despite the activity in combination with irradiation in head and neck cancer, cetuximab combined with chemotherapy and irradiation may have a negative impact on the pathologic complete response in rectal cancer.47 Hypotheses for this include the up-regulation of cyclin-dependent kinase p27, G1 cell cycle arrest induced by cetuximab, and the redundancy of the EGFR pathway.
Another tyrosine kinase inhibitor, imatinib, was designed to target the BCR/ABL mutation that causes chronic myelogenous leukemia. Imatinib produces long-term disease control, with normalization of both blood counts and cytogenetics, in the chronic phases of the disease.48 Newer inhibitors, norlotinib and dasatinib, kill chronic myelogenous leukemia cells with mutations that confer resistance to imatinib and provide alternative therapy for drug-resistant patients. Imatinib was also found to inhibit the KIT (CD117) receptor, and it has proved to be a powerful agent in the management of gastrointestinal stromal tumors. Overexpression of KIT is not the sole predictor of response to imatinib, because this agent has been shown to be ineffective in small cell lung cancer, a tumor that overexpresses KIT. Identification of specific activating mutations within the KIT gene predicts for response in patients with gastrointestinal stromal tumors49 (see Table 8-3). The multitargeted tyrosine kinase inhibitor sunitinib has demonstrated benefit in imatinib-refractory patients, even in those with mutations that are typically associated with a low response rate to imatinib.50
Other regulatory pathways have attracted the interest of drug discovery programs. The phosphatidylinositol-3-kinase (PI3K) pathway activates proliferation and down-regulates the activity of the intrinsic pathway of apoptosis. Inhibitors of various components of this pathway, including PI3K, AKT, mammalian target of rapamycin (mTOR), and IGF-1R, display antitumor activity. There are multiple agents in development and several that target the PI3K-AKT-mTOR pathway, have been approved. The drugs furthest along in the development process are the mTOR inhibitors. Temsirolimus (CCI779) is a synthetic rapamycin ester that received FDA approval for previously untreated patients with high-risk advanced renal cell carcinoma. In a randomized trial of temsirolimus, interferon, or the combination of both agents, progression-free survival rates favored temsirolimus over interferon (3.8 months vs. 1.9 months; p <.001), as did improvement in overall survival rates (10.9 months for temsirolimus, 7.3 months for interferon, and 8.4 months for the combination regimen).51 Everolimus (RAD001) is another mTOR inhibitor that is FDA approved for the treatment of renal cell carcinoma in patients refractory to prior targeted therapies; everolimus improved progression-free survival rates compared with placebo (4 months vs. 1.9 months; hazard ratio [HR], 0.3; p <.0001).52 Everolimus has also demonstrated activity in carcinoid and pancreatic neuroendocrine tumors.53,54 Development of agents targeting PI3K and AKT has been more challenging, and these agents remain under development. Anti-IGF1-R antibodies have notable activity against Ewing’s sarcoma and are progressing through clinical evaluation.
Activating translocations of the anaplastic large-cell kinase gene (ALK) have been found in about 5% of a Western population of patients with NSCLC.55 In a phase I/II trial of the MET-ALK inhibitor PF-02341066, patients possessing the ALK fusion protein exhibited a surprising 53% clinical response rate, with an additional 20% of patients having stable disease for 3 months or more.56 This agent will be evaluated in a phase III trial in patients with NSCLC possessing the ALK fusion protein.
In 2003, the FDA approved bortezomib, a proteosome inhibitor, for patients who have progressed while receiving prior therapy for multiple myeloma. Although the exact mechanism of action for this drug is unclear, it acts in part by down-regulating nuclear factor kappa B (NFκB), a potent survival pathway for cancer cells.57 It is currently approved as part of first-line therapy for multiple myeloma in combination with melphalan and prednisone and as second-line therapy as a single agent for mantle cell lymphoma. It is also being tested in other hematologic malignancies and in solid tumors, either as a single agent or in combination with cytotoxic chemotherapy and radiation therapy.