Fig. 3.1
Model showing the progress of disease and potential stages of predictability and prevention of T1D relating it to the loss of β-cell mass and time. Modified from Eisenbarth (1986)
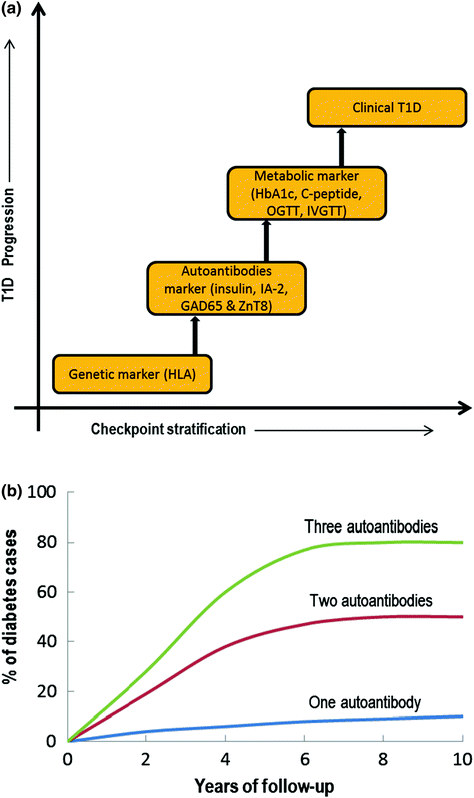
Fig. 3.2
a Checkpoint classification based on the biomarkers used to indicate T1D progression, b Graphical representation correlating autoantibody numbers and % cases of first-degree relatives with T1D progression based on several longitudinal studies (OGTT = Oral glucose tolerance test; IVGTT = Intravenous glucose tolerance test; HbA1C = Glycated hemoglobin). Modified from Notkins and Lernmark (2001)
As described above, in addition to genetic susceptibility, various other trigger agents are involved in the initiation and progression of autoimmune T1D. For example, the presence of autoantibodies is often observed years before the clinical onset of T1D (Leslie et al. 2001). Reports suggest that autoantibodies may be transferred via the placenta or develop after birth, but this is rarely diagnosed before the age of 6 months (Rubio-Cabezas et al. 2009). More than two dozen autoantigen-related autoantibodies have been implicated although four major islet autoantibodies have drawn significant attention: insulin (or proinsulin or IAA) (Palmer et al. 1983), GAD isoforms (65 and 67) (Baekkeskov et al. 1990), tyrosine phosphatase-related islet antigens (IA-2 or ICA512) (Rabin et al. 1994), and the most recently identified zinc transporter (ZnT8) (Wenzlau et al. 2007). However, the positive detection of any one of the autoantibodies mentioned only imparts a marginal increase in the risk of developing T1D and to date it has not been possible to reliably predict progression to clinical disease although incremental increases in disease risk were found in individuals who were positive for two, three, or more autoantibodies (Fig. 3.2b) (Verge et al. 1996). Relative risk was also found to vary dependant on autoantibody type with some reports emerging that the most influencing antibody is insulin, as identified in NOD mice (where diabetes was inhibited by single amino acid mutation in insulin peptide 9-23) and as the only β-cell-specific autoantibody identified in T1D (Nakayama et al. 2005). In contrast, several other studies support GAD65 and IA-2 as high risk-associated autoantibodies in humans. Nonetheless, higher numbers of autoantibodies, high titre, and affinity for epitopes to various antigens govern the degree of risk associated to any particular antigen group (Verge et al. 1998).
In the late 1990s, when screening using recombinant antigen assay methods were common place, about 5–10 % of the adult T2D population had autoantibodies for GAD65 and a slightly lower percentage (2–4 %), tested positive for IA-2. If this is the case, it could be inferred that many of the patients who were classified as T2D sufferers, actually either had T1D or a combination of T1D and T2D (Turner et al. 1997; Hawa et al. 2000). Based on this hypothesis, it could further be concluded that the number of T1D patients would actually be double than the current estimates. The current approach to diagnosis includes a stepwise decision tree to identify individuals at high risk of developing T1D (see Fig. 3.2a). Initially, genotyping for susceptible HLA II genes and patients with a family history is tested. Individuals or children (≥age 1 year) who warrant sufficient HLA susceptibility in the first stage are then measured for their autoantibody levels. Additional genotyping in autoantibody positive individuals helps exclude the one with protective HLA II genes. As a final check, often insulin-secreting capacity is measured as an indicator of β-cell function or IVGTT, OGTT, and C-peptide levels are also measured (Srikanta et al. 1985).
Cumulative evidence indicates the multifactorial nature of T1D due to associated complex immunology, and this necessitates the use of a combinatorial screening method to identify the individuals at risk of developing T1D. In addition, continual efforts toward extracting reliable, specific biomarkers have led to the identification of some promising serum proteins, namely cytokines and chemokines (Wasserfall and Atkinson 2006). Cytokines have been proposed as one of the major inducers of β-cell damage especially the type 1 cytokines (Th1) such as interferon gamma (IFN-γ), tumor necrosis factor-α (TNF-α), and IL-2 and they are considered ‘high risk’ with approximately 40 % of patients going on to develop T1D within 5 years (Ryden et al. 2009). However, a lack of specificity of cytokines for T1D, their varied level of expression, and trace amount poses serious analytical challenges for their quantification, when placed against a background of large numbers of non-relevant proteins (Borges et al. 2010; Carey et al. 2010). Despite considerable global efforts and investment progress in the area of T1D disease prediction has progressed at a painfully slow pace, with researchers keenly anticipating that advancements in proteomics and metabolomics will serve as a future platform for the identification of reliable, specific biomarkers for T1D in the coming era (Cassiday 2008).
3.1.1 Pathophysiology of Type 1 Diabetes: What Goes Awry?
3.1.1.1 T-Cells: A Double Edge Sword
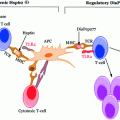
Given that T1D is primarily an autoimmune disease, the mechanisms of which are poorly understood, the actions of a handful of antigens such as insulin, GAD isoforms (GAD65 & 67), IA-2, and ZnT8 are known to transform into autoantigens. In individuals of high susceptibility the pancreatic Islet of Langerhans are primarily infiltrated by APCs such as DCs and Mϕs. Concomitantly or shortly thereafter autoreactive CD4+ and CD8+ T-cells can be readily detected surrounding the islets (known as peri-insulitis, see Fig. 3.3) (Turley et al. 2003) as highlighted in a study by Haskins et al., where it was demonstrated that progression of autoimmune diabetes occurred in NOD mice that received adoptive transfer of CD4+-specific T-cells from NOD mice compared to placebo (Haskins and McDuffie 1990; Wong et al. 1996). Due to ethical concerns, only recently the examination of pancreatic biopsy in prediabetic patients been possible; this makes its feasible to study the composition of any lesions (insulitis).
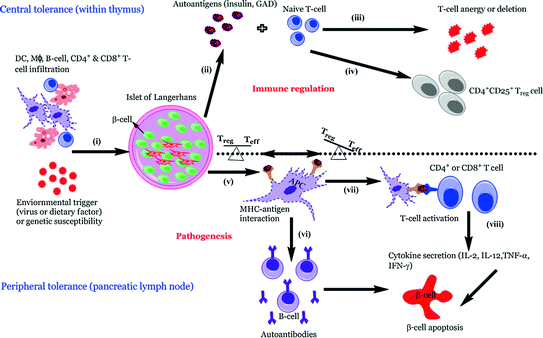
Fig. 3.3




Pathogenetic model of T1D. In individuals of high susceptibility, the pancreatic Islet of Langerhans are primarily infiltrated by APCs such as DCs and Mϕs followed by autoreactive CD4+ and CD8+ T-cells which can be detected surrounding the islets (known as peri-insulitis) (i). This islet damage is followed by autoantigen release (ii) and while on one hand thymic immune regulation leads to active tolerance by deleting T-cells (iii) or inducing protective, regulatory T-cells (CD4+ CD25+ Treg) (iv) on other hand its failure leads to escape of autoreactive T-cells into peripheral pancreatic lymph nodes, inducing an autoimmune response. In order to prevent this autoimmune response it is essential to maintain a balance between the level of Treg (‘good cells’) and effector Teff (‘bad cells’) T-cells. The released autoantigens are taken up by the APCs (mainly DCs) (v) and from here, there is a diversion in the fate of APCs. One path leads to APCs stimulating B-lymphocytes, which produces autoantibodies toward the autoantigens (vi) (Lennon et al. 2009) and the role of these B-lymphocytes is briefly discussed below. Another possibility is the conversion of the autoantigens into peptides followed by pMHC–peptide complex formation. Further, these complexes are presented onto CD4+ or CD8+ (Teff cells) TCRs which activate T-cells (vii). This causes the release of proinflammatory cytokines and chemokines (IL-2, TNF-α, and CTL) (viii) and ultimately leads to β-cell apoptosis resulting in eventual loss of insulin production once a critical mass of β-cells have been eliminated (Ounissi-Benkalha and Polychronakos 2008; Atkinson et al. 2011)
Related posts:

Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
