Fig. 17.1 Neuroendocrine cell hyperplasia: Bronchiolar epithelium is undermined by a uniform proliferation of round to oval neuroendocrine cells, without stromal invasion
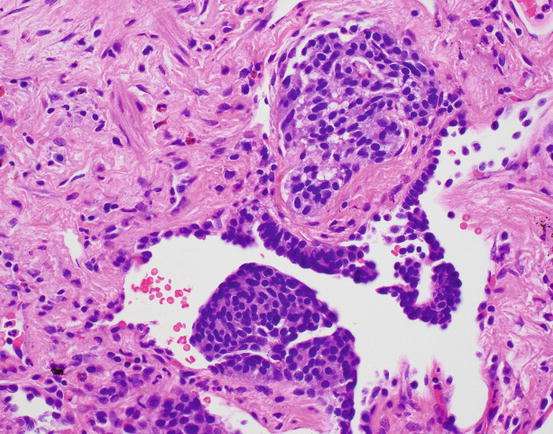
Fig. 17.2 Neuroendocrine cell hyperplasia. Proliferations of cells can be exuberant, causing polypoid projections into the airway lumen
Carcinoid tumors are characterized by an absence of 3p loss, normal FHIT, low telomerase, and MEN1 mutations. Molecular alterations in these tumors are different than those of adenocarcinomas and squamous carcinomas. While 11q 13 allelic imbalance is seen in carcinoids, it is not present in tumorlets [3]. Developing neural transcription factors are seen in neuroendocrine proliferations but not in normal neuroendocrine cells. These include TTF1, ASCL1, and POU3F2. VGF peptides increase in hyperplasia and neoplasia [4].
There are mouse models of small cell carcinoma that attempt to shed some light on potential cells of origin for neuroendocrine tumors. An animal model inducing p53 loss and RB loss in epithelial cells induced small cell carcinoma expressing ASCL1 [5]. By using adenovirus cre recombinase delivery into the airway of mice that are engineered to induce loss of p53 and RB in different types of epithelial cells, the loss of P53 and RB directed toward neuroendocrine cells led to small cell carcinoma. Only rare neuroendocrine tumors were derived from targeting type 2 pneumocytes than produce surfactant protein C [6].
Atypical Adenomatous Hyperplasia
Atypical adenomatous hyperplasia (AAH) is a proliferation of type 2 pneumocytes that is generally small and incidentally discovered. While a size criterion has not been absolutely set, these are usually under 5.0 mm. They are characterized by a proliferation of pneumocytes with mild to moderate atypia, but importantly these proliferations show variability from cell to cell and leave residual type 1 pneumocytes along the alveolar walls, seen as gaps in the proliferation (Fig. 17.3). Alveolar walls may be mildly thickened, but they are not invaded. Such lesions were described by Roberta Miller in 1988 as possible precursor lesions, akin to adenomas of the colon [7].
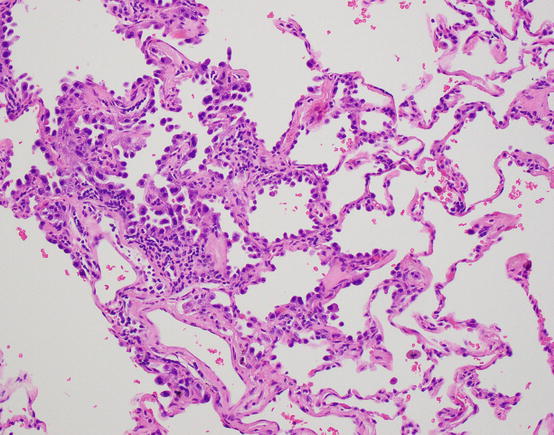
Fig. 17.3 Atypical adenomatous hyperplasia. Alveolar lining proliferations of hobnail and atypical pneumocytes, with cell to cell variability and gaps of residual type 1 cells
These AAH lesions are frequently found in adjacent lung tissue of patients with pulmonary adenocarcinoma but also can be found in lung tissues removed for nonneoplastic indications [8]. AAH is found more frequently in lungs from patients with adenocarcinoma (23.2%) and in women with adenocarcinoma (30.2%). AAH is seen more frequently in patients with multiple carcinomas, usually multiple adenocarcinomas [9].
An important question in the study of precursor lesions of neoplasia is their clonality . AAH lesions in women were studied, and their pattern of X-inactivation supports the clonality of these lesions [10]. In addition, the evidence supports that multiple AAH are independent foci.
These observations point to the conclusion that atypical adenomatous hyperplasia is neoplastic and has a similar pathogenesis as adenocarcinoma. Its morphologic resemblance to early adenocarcinoma such as adenocarcinoma in situ and minimally invasive adenocarcinoma, albeit smaller and with a less uniform proliferation, is also indirect evidence of AAH as a precursor lesion to subtypes of adenocarcinoma. However, the percentage of AAH lesions that progress to adenocarcinoma is unknown; in other words, it is not known whether this is an obligate precursor to adenocarcinoma or a non-obligate one. The latter seems more likely given the number of such lesions that can be present in an individual lung resection. The latency of progression is also not known. Although authors have described a connection with TTF1-positive peripheral adenocarcinoma and have coined the term terminal respiratory unit type adenocarcinoma, what proportion of adenocarcinomas arises from a progression of AAH to adenocarcinoma in situ is also speculative. The nonterminal respiratory unit adenocarcinoma has a less well-defined precursor. It is likely that a non-lepidic precursor exists, but its histologic appearance remains poorly described [11, 12].
Loss of Heterozygosity (LOH) Data and Loss of Tumor Suppressor Activity
Loss of heterozygosity has been reported in atypical adenomatous hyperplasia at several loci, and these are seen at increased frequency in adenocarcinoma . For example, in a study of 18 AAH and 17 AdCa, loss of heterozygosity at a 16p locus was seen in 22% of atypical adenomatous hyperplasia while in 35% of adenocarcinomas and at a 9q locus in 28% of AAH and in 41% of AdCa. The locus in 9q included the TSC1 gene [13].
Loss in 3p is a commonly reported finding in 10–15% of AAH [14–16]. However, loss of FHIT does not appear to occur in AAH (see “Squamous Dysplasia” section), suggesting that the 3p loss involves a different locus. Losses in 9p in roughly 13% of cases and in 17p in 5% of cases have been described. Allele-specific LOH at 9p locus was seen in preinvasive lesions and identified at an increasing rate in invasive carcinoma [17]. The likely target of this LOH is CDKN2a (p16), but other pathways of silencing may also be in play (see section on “Promoter Hypermethylation”). In addition to 3p, 9p, and 17p, LOH at 5q, 7p, and 10q in AAH is described [18]. Overall, these studies suggest that there are common regions of loss in AAH and adenocarcinoma and that the regions of loss increase during progression.
P53 Mutations
P53 IHC was studied in AAH, with p53 overexpression in 17–53% of cases [19, 20]. However p53 mutation rate may be lower than IHC immunoreactivity would suggest. In one series, no p53 mutations were seen [16], also in support of the overestimation of p53 mutation by IHC. In addition, a demonstration of increasing rate of p53 mutation from AAH to adenocarcinoma in situ and invasive adenocarcinoma with sclerosis has been described along with increasing rate of 17p LOH in this progression, the chromosomal region containing p53.
Epidermal Growth Factor Receptor (EGFR) and Kirsten Rat Sarcoma Viral Oncogene Homolog (KRAS) Mutations
KRAS
There is conflicting data on KRAS mutations in AAH. In one series of 19 AAH, no KRAS mutations were reported [18]. In an early study of AAH, KRAS mutations were found in 39% of lesions [21], with transversion mutations similar to those seen in cigarette smokers. This was also reported in a separate study of 32 AAH lesions in which 16% harbored KRAS transversion mutations [22].
Some of this data suggests that AAH lesions harboring KRAS mutations are the result of cigarette smoking, so different reported rates could reflect study populations with different smoking rates. Within the lung tissue exposed to cigarette smoke, separate independent KRAS mutations may be induced. For example, in six patients with AAH, 2 of 6 patients with multifocal lesions [23] harbored KRAS mutations, overall in 50% of their AAH lesions. Of note, specific KRAS mutations in AAH did not match those in adenocarcinomas from the same patient [21].
EGFR Mutations
Early studies of EGFR mutation in AAH also showed variable results and included detection in 2 of 7 (28%) AAH [24] in one series and 0 of 5 AAH in another [25]. In a series of 18 AAH, 3 of 18 (17%) showed EGFR mutations with 2 of 18 cases (11%) with KRAS mutations [26]. As experience was gained in larger series, 17 of 54 (32%) AAH showed EGFR mutation [27], with 10 cases of exon 19 deletion and 7 with exon 21 (L858R) mutations. In that set, KRAS mutations were seen in only 1 of 49 AAH (2%). The same authors [28] found a high rate of EGFR mutations in their non-mucinous lepidic pattern containing adenocarcinomas in 64 out of 82 (78%), while KRAS was seen in only 2 of 82 (2.4%). It may be that the low KRAS rate in part reflects an East Asian, nonsmoking population as the explanation for the relatively low KRAS mutation rate.
In an interesting study of adenocarcinoma and their precursors, among 40 AAH lesions, 33% harbored KRAS mutations, while 25% had EGFR mutations, predominantly exon 21 L858R missense point mutations. In the same study, only 12% of adenocarcinoma in situ (AIS) and 12% of invasive adenocarcinoma harbored KRAS mutations, while 36% of AIS and 56% of adenocarcinoma had EGFR mutations, with a relatively balanced distribution of exon 19 and exon 21 mutations [29]. This group noted an increased number of KRAS mutations in smokers. Another series showed a rate of EGFR mutation of 3% in AAH and 11% in AIS, with KRAS mutations in 27% of AAH and 17% of AIS [30]. These observations suggest that KRAS mutations may occur at a higher rate in AAH than adenocarcinoma in these East Asian populations and that the opposite is true in EGFR mutation. One possibility is that EGFR mutations, when they occur in precursor lesions, are more likely to be obligate precursors to adenocarcinoma or cause more rapid progression to adenocarcinoma, and therefore their rate in precursor lesions appears low.
Looking beyond AAH among AIS preinvasive and early invasive lesions, Soh et al. showed EGFR mutations in 1 of 4 AAH (25%) and a rate of 36, 47, and 50% using the schema of Noguchi A, B, and C, respectively. In the same series, KRAS mutations were seen in 10, 6.7, and 9% in these groups. In a series of 17 AIS lesions (noninvasive BAC) in an East Asian cohort, 88% had EGFR mutations [28] (adenocarcinoma in situ). In a Western series, among 18 cases of non-mucinous AIS and minimally invasive adenocarcinoma, 7 of 18 were associated with EGFR mutation with 1 of 18 harboring KRAS mutation [31].
EGFR Copy Number Gain
Copy number gains in EGFR were seen in AAH with increased rate and level of gains in adenocarcinoma [32]. In the series of Soh et al., EGFR copy number increase occurred during the progression [33] from Noguchi A, B, and C of 5.3, 13, and 32%, respectively. It has been suggested that EGFR amplification could represent a marker of progression.
Measures of Cell Cycle Activity
An examination of cell cycle -related markers in AAH showed decreased CDKN2A (P16) expression with concomitant increase in cyclin D in 55% of cases. However, KI67 was low in these lesions [34]. While proliferation was higher in adenocarcinoma in situ than in AAH, invasive adenocarcinomas showed the highest proliferation index; this observation is somewhat limited by changes in terminology that have occurred since these studies were conducted [14, 20].
The inhibition of p21CIP1 protein results in activation of a pathway of cell cycle initiation and progression, that is, the RB/CDK/cyclin D1 axis is activated when p21 levels are low. However, this may be a later event in carcinogenesis as low p21 is seen in invasive adenocarcinoma [35]. Inhibitors of CDK2 such as p27Kip1 may be actively degraded by JAB1, and higher levels of JAB1 were identified in AAH lesions when compared to normal pneumocytes [36], with increasing frequency of expression from AAH (36%) to adenocarcinoma (54%).
Immunohistochemistry Studies
In an IHC study designed to examine loci related to acquisition of invasion, FHIT was decreased in invasive adenocarcinoma but retained in AAH. CD44v6 and TIMP2 also showed a decrease in the progression from AAH to invasive carcinoma [37]. Destruction of type IV collagen and MMP2 expression have also been associated with invasion; preservation of type IV collagen was seen in all AAH cases with some cases showing MMP2 without its activating enzyme [38]. MMP2 expression was seen to increase from AAH and adenocarcinoma in situ to invasive adenocarcinoma with central scar formation [39].
Gene Expression Studies
Gene expression studies of lung adenocarcinoma [40, 41] have shown differences between preinvasive adenocarcinomas and invasive adenocarcinoma that contain a lepidic component (in some studies, termed terminal respiratory unit B and A type, respectively). In the series by Takeuchi et al., the noninvasive and focally invasive adenocarcinomas were in the TRU-b group, with EGFR mutations in a subset of these cases, but no p53 or KRAS mutations. Their expression was characterized by gene ontologies related to cellular differentiation. Gene expression studies have also implicated loss of TGFBR2 expression, the receptor for TGF-B, in the acquisition of invasion in adenocarcinoma, and coordinated epithelial cell activation by oncogenic KRAS and loss of TGFBR2 in a mouse model resulted in invasion and metastasis [31].
Other Molecular Alterations
In an examination of DNA methylation in AAH, changes in DNA at loci related to CDKN2a exon 2 and PTPRN2 were seen [42]. Earlier studies also identified CDKN2a promoter hypermethylation in AAH [43]. In the transition from AAH to AIS, further alterations were seen in HOXA1, HOXA11, NEUROD1, NEUROD2, and TMEFF2 and from AIS to adenocarcinoma in CDH13, CDX2, OPCML, SFRP1, TWIST1, and RASSF1.
In one study using mass spectroscopy, differences were noted from AAH to carcinoma, although no specific protein was identified characteristic of that transition [44]. Increase in human telomerase RNA component (hTERC) and telomerase reverse transcriptase (hTERT) mRNA expression by in situ hybridization was described in AAH with increasing rates in lepidic pattern containing adenocarcinomas [45]. In situ hybridization for telomeric repeat-binding factor 1 (TERF1) also showed increase in a similar fashion [46].
Non-AAH Proliferations
In a search for non-AAH precursors of adenocarcinoma, Ullman and colleagues [47] examined airway glandular dysplasia by comparative genomic hybridization and found gain in 1q, 17, 19q, and 20q in these putative precursor lesions, with losses at 3p, 9, 13, and 14. However further study of such lesions remains to be pursued.
Congenital pulmonary airway malformation type 1 can be associated with mucin producing cellular proliferations that resemble adult-type mucinous adenocarcinoma (Fig. 17.4). These can have lepidic mucinous growth and can be both within and outside the cystic lesions [48, 49]. Of interest is the description of oncogenic KRAS mutations in these lesions supporting their neoplastic nature. Additionally, LOH at FHIT, Rb, and CDKN2A have been shown as well as gains in chromosomes 2 and 4. Their biological relationship to adult mucinous adenocarcinoma, while linked by these KRAS mutations and other alterations, is unknown. While more indolent than their adult counterpart, extrapulmonary spread has been reported [50].
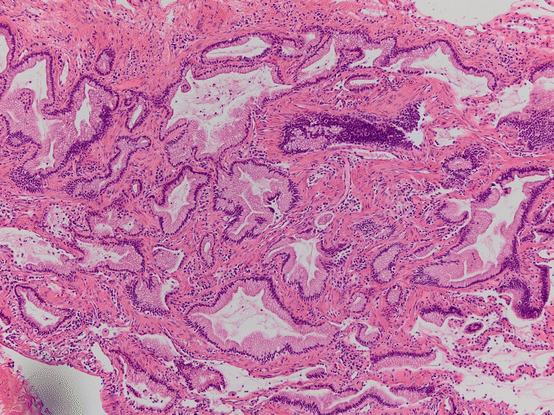
Fig. 17.4 Mucinous proliferation in CPAM. Bland mucinous proliferations morphologically mucinous adenocarcinoma are seen lining pre-existing airways but also with probably stromal invasion
Tumors resembling adenocarcinoma in situ, non-mucinous type, have been described in children, adolescents, and young adults, who are nonsmokers and frequently have a history of treatment for an unrelated malignancy. Only seven reported cases were tested for EGFR and KRAS mutations, with 2 of 7 and 1 of 7 positive, respectively. Copy number analysis revealed polysomy 17, as well as other copy number gains [51–53].
Peripheral glandular papillomas , recently renamed ciliated muconodular papillary tumors (Figs. 17.5 and 17.6), have been studied for mutations and found to have BRAF V600E and EGFR exon 19 deletions (specifically E746-T751/S752V mutations) [54]. Recently, two cases were reported with ALK translocations [55, 56]. These alterations are of interest as they are all encountered in lung adenocarcinoma, but to date, malignant transformation of peripheral glandular papillomas or ciliated muconodular papillary tumors has not been reported.
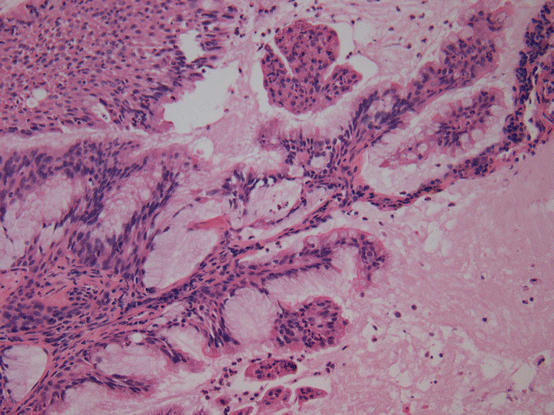
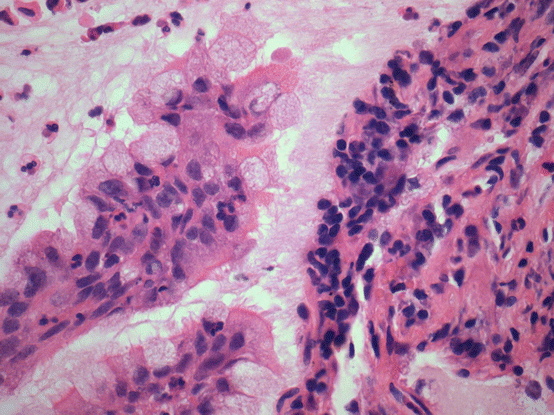
Fig. 17.5 Ciliated muconodular papillary tumor/peripheral glandular papilloma. Mcuin filled airspaces are lined by an admixture of mucinous cells, including goblet cells, and ciliated cells
Fig. 17.6 High magnification highlights ciliated cells adjacent to mucinous cells
Squamous Dysplasia and Squamous Carcinoma In Situ
The identification of early preinvasive squamous lesions, ranging from metaplasias, dysplasias (both low and high grades), and carcinoma in situ, has been enhanced by white light bronchoscopic examination, with special tools such as autofluorescence bronchoscopy [57] and narrow band imaging [58] providing enhanced detection. These lesions are associated with cigarette smoking and often heavy tobacco exposure [59]. Lesions can be seen throughout the airways of affected individuals and can be flat or polypoid [60]. They can be erythematous or exhibit leukoplakia. Regression of low-grade dysplasia, the time to progression to invasion, and the frequency by which lesions progress to invasive squamous carcinoma are not known.
The histologic diagnosis of these lesions depends on morphologic assessment of squamous differentiation in metaplasia, characterized by stratified squamous epithelium rather than pseudostratified columnar ciliated epithelium typical of the normal large airway. Within squamous metaplasia, mild nuclear atypia and preservation of surface maturation are typical of low-grade dysplasia, while a thickened epithelium showing dysmaturation with mitotically active cells showing marked nuclear atypia is a characteristic of high-grade dysplasia. Full-thickness nuclear atypia, mitotic activity, disarray, and complete lack of maturation are features of carcinoma in situ (Fig. 17.7). Multiple carcinoma in situ lesions in the same patient harbor the same p53 mutations, suggesting common clonal origin and cellular migration [61].
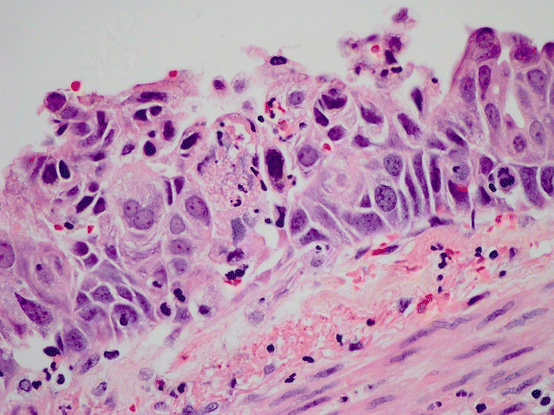
Fig. 17.7 Squamous carcinoma in situ. Squamous epithelium replaces the normal respiratory epithelium, and marked atypia and full thickness dysmaturation are seen. In addition mitotic activity and apoptosis is present
Loss of Heterozygosity and Stepwise Progression of Dysplasia
Allele-specific deletions in 3p were identified in squamous hyperplasia, dysplasia, and squamous carcinoma in situ [62]. Progressive LOH in 3p is seen from low-grade to high-grade dysplasia and is also frequent in carcinoma; both the rate and extent of LOH were seen with increasing grade [63]. LOH was found showing allele-specific losses in 3p, 5q, 9p, 13q, and 17p, with the 3p losses representing earlier events, followed by 9p losses and then 5q, 13q, and 17p [64]. The 3p allele-specific deletions are seen in 76% of hyperplasias [62]. LOH at 9p are also allele specific and start to occur in the squamous hyperplasia histology [62]. This progressive loss includes region 3p21; losses were also described in 5q21 and 9p21 [65]. Progressive LOH at 3p, 5q, 9p, and 17p mutations were seen in high-grade dysplasia [66].
Loss of 3p including the tumor suppressor gene fragile histidine triad (FHIT) was proposed to be important in squamous carcinogenesis [63]. The function of FHIT is complex and includes effects on cell cycle, invasion, and apoptosis. The losses at 9p are likely targeting CDKN2a (p16) and losses at 17p targeting p53.
Acquisition of Invasion
IHC studies have examined MMP levels with decreased MMP and increased MMP3 and 9 from dysplasia to carcinoma in situ [67]. P53 IHC was seen to increase from high-grade dysplasia to carcinoma in situ and microinvasive carcinoma [68]. IHC for EGFR and p53 IHC were seen to increase from dysplasia to carcinoma [69].
Telomerase Activity
Telomerase activity was shown to increase from metaplasia to dysplasia to carcinoma [70]. This was also reported by Lantuejoul and colleagues, and HTERT level was correlated with p53 loss, increased BCL2-to-BAX ratio, and Ki67 increase. Additionally, increased telomere length was seen with carcinoma in situ and squamous carcinoma [71]. Telomere length is shortest at the squamous metaplasia step and increases from dysplasia to squamous carcinoma. This is associated with increase in telomere repeat factors 1 and 3 (TRF1 and TRF2). Other DNA damage response proteins are also increased, including p-ATM, p-CHK2, and p-H2AX.
Cell Cycle Control
Squamous dysplasias were shown to have CDKN2a (p16) loss and gains in cyclin D1 and cyclin E1, but no loss in Rb [72]. This was also reported by Lantuejoul and colleagues, with CDKN2a (p16) loss and cyclin D1 gain [71]. Loss of 9p21 and 1p36 in dysplasia and carcinoma [73] is described. Abnormalities in retinoblastoma pathway were also found [74].
Loss of CDKN2a (p16) by promoter hypermethylation and polycomb complex component BMI-1 overexpression was seen in cell proliferation with severe squamous dysplasia, as measured by MIB-1 [75]. CDKN2a (p16) promoter hypermethylation in smokers was thought to be an early event [76], and CDKN2a (p16) hypermethylation was seen in 17% of hyperplasia, 24% of squamous metaplasia, and 50–75% of carcinoma in situ and invasive squamous carcinoma [77]. The silencing of CDKN2a (p16) and LOH at that locus are mechanisms by which cell cycle control is progressively lost along the RB/CDK4/cyclin D1 axis.
Antiapoptotic Effects
PIK3CA copy number from low-grade to moderate dysplasia to carcinoma in situ was associated with phosphorylated AKT in high-grade dysplasia. It was postulated that prevention of cell death mediated by the phospho-AKT pathway was the result of PIK3CA copy number increase [78]. The antiapoptotic pathway components including p-AKT, p65 RELA, and cIAP-2 were increased in the progression from precursors to bronchial squamous carcinoma [79].
TP53 Loss
Loss of TP53 is a critical event in human malignancy as loss of TP53 has an effect on apoptosis as well as on cell cycle. TP53 mutations, however, increased in frequency from dysplasia to CIS [80]. Mutations in TP53 are seen in high-grade dysplasias [66], and LOH in 17p also targets TP53 as previously noted. The loss of TP53 is likely related to cigarette smoking and may already be detectable in normal-appearing epithelium [81]. The importance of the p53 pathway was further underscored by the work of Masscaux et al. in which it was noted that MDM2 increases were seen from metaplasia to dysplasia. These increases were associated with p14ARF loss or nucleolar localization in lesions with MDM2 increase [82]. The role of miRNA has also been explored with subsequent p53-associated targeting. Reduced Mir32 and Mir34C were seen with progression from normal to dysplasia to carcinoma. Mir15A was downregulated in early dysplasia and is important in regulating apoptosis [83].
EGFR and KRAS Mutations
EGFR and RAS mutations were found to be rare in bronchial squamous dysplasia [84].
Copy Number Alterations and SOX2
Single-nucleotide polymorphism arrays showed multiple regions of chromosomal gains and losses in preneoplastic squamous lesions. These copy number alterations included 3p, 5p, 8p, 9p, and 13q. In addition, loss of RNF20 and SSBP2 and gain of RASGRP3 were also lesions seen in squamous carcinoma, associated with changes in 9q31.1, 2p22.3, and 5q14.1.
Aneuploidy increases from high-grade dysplasia to carcinoma [68] including copy number changes in 5p15.2, chromosome 6, 7p12 (EGFR), and 8p24 (myc) frequent in dysplasia [85]; these changes increased in frequency from low-grade to high-grade dysplasia and to squamous carcinoma.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


