Maintenance of the tonicity of extracellular fluids within a very narrow range is crucial for proper cell function because extracellular osmolality regulates cell shape, as well as intracellular concentrations of ions and other osmolytes.1,2 Furthermore, proper extracellular ionic concentrations are necessary for the correct function of ion channels, action potentials, and other modes of intercellular communication. Normal blood tonicity is maintained over a 10-fold variation in water intake by a coordinated interaction among the vasopressin, thirst, and renal systems. Dysfunction in any of these systems can result in abnormal regulation of blood osmolality, which, if not properly recognized and treated, may cause life-threatening hyperosmolality or hypo-osmolality.
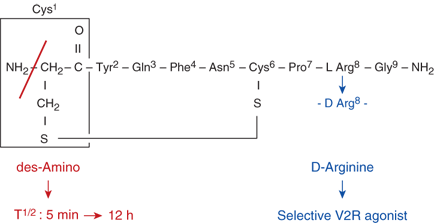
The control of plasma osmolality and intravascular volume involves a complex integration of endocrine, neural, and paracrine pathways. Plasma osmolality is regulated principally via vasopressin release from the posterior pituitary, or neurohypophysis, while volume homeostasis is determined largely through the action of the renin-angiotensin-aldosterone system, with contributions from both vasopressin and the natriuretic peptide family. The nine-amino acid vasopressin (also known as antidiuretic hormone [ADH] and arginine vasopressin [AVP]) molecule consists of a six-amino acid disulfide ring plus a three-amino acid tail, with amidation of the carboxy-terminus (Figure 8-1). Vasopressin has both antidiuretic and pressor activities, actions caused by the hormone binding to different receptors, as discussed subsequently. A synthetic analog of vasopressin, DDAVP (desamino-D-arginine vasopressin, desmopressin) with twice the antidiuretic potency and 100 times the duration of action of vasopressin, and no pressor activity, is routinely used to treat vasopressin-deficient patients (see Figure 8-1).3
Figure 8-1
Molecular structure of vasopressin and desmopressin. Substitution of the eighth amino acid l-arginine of the nonapeptide vasopressin with D-arginine confers selective agonist activity on the V2 receptors. Removal of the amino group from the first amino acid cysteine prolongs the half-life significantly because vasopressinase attacks vasopressin from this end of the molecule.
Vasopressin is synthesized in hypothalamic paraventricular and supraoptic magnocellular neurons, whose axons transport the hormone to the posterior pituitary, its primary site of storage and from where it is released into the systemic circulation (Figure 8-2). The bilaterally paired hypothalamic paraventricular and supraoptic nuclei are separated from one another by relatively large distances (∼1 cm). Their axons course caudally and converge at the infundibulum, before terminating at different levels within the pituitary stalk and posterior pituitary gland.4 This anatomy has important clinical implications, as will be discussed. Vasopressin is also synthesized in the parvocellular neurons of the paraventricular nucleus (PVN), where it may have a role in regulating the hypothalamic-pituitary-adrenal axis, and in the hypothalamic suprachiasmatic nucleus, where it may participate in the generation of circadian rhythms.
Figure 8-2
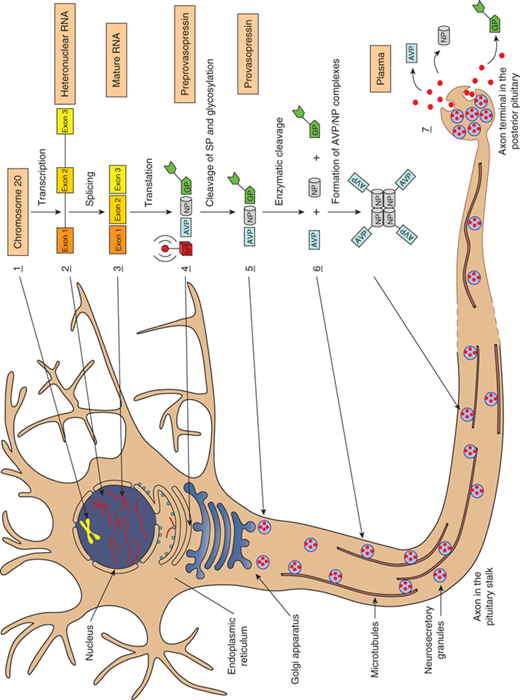
Steps in the synthesis and release of vasopressin. 1. Nucleus: Transcription of the vasopressin gene (chromosome 20) to heteronuclear RNA. 2. Nucleus: Synthesis of the mature RNA by removal of the introns and the splicing of exons A, B, and C, and its subsequent passage into the cytoplasm. Attachment of the mRNA to ribosomes in the endoplasmic reticulum (ER). 3. Endoplasmic reticulum (ER): Translation of the exons to preprovasopressin. Exon 1 encodes the 19-amino acid (AA) signal peptide (SP), the 9-AA vasopressin (AVP), and the aminoterminal of the 93-95-AA neurophysin (NP); exon 2 encodes the middle portion of neurophysin; and exon 3 encodes the carboxy-terminal of neurophysin and a 39-AA glycopeptide (GP). 4. Endoplasmic reticulum (ER): Glycosylation of the glycopeptide portion of the preprovasopressin and cleavage of the signal peptide. 5. Golgi complex: Entry of provasopressin into the Golgi complex and its packaging into neurosecretory granules. Attachment to and the subsequent transport of the granules along microtubules to the site of storage in the posterior pituitary gland. 6. Neurosecretory granules: Enzymatic cleavage of provasopressin to vasopressin, neurophysin, and glycopeptide during the transport within the acidic granules. Amidation of vasopressin, formation of complexes consisting of one vasopressin molecule with either a dimer or a tetramer of neurophysin. 7. Neurosecretory granules: Fusion of the granule with the plasma membrane as a result of an action potential and the release of vasopressin, neurophysin, and the glycopeptide into the extracellular space and plasma. (Modified with permission from Alan Robinson, MD, UCLA.)
During its synthesis and perhaps after secretion into the bloodstream, vasopressin is bound to neurophysin, a 10,000-Da (dalton) molecular weight protein, which may function to protect vasopressin against degradation during intracellular storage or to promote more efficient packaging or posttranslational processing of vasopressin within secretory granules (Figure 8-3). Vasopressin and neurophysin are made from a common gene that consists of three exons. Exon 1 encodes the signal and vasopressin peptides and the aminoterminal 9 amino acids of neurophysin, exon 2 encodes the majority of neurophysin, and exon 3 codes for the carboxy-terminal end of neurophysin followed by an additional 39-amino acid-long glycopeptide.
Figure 8-3
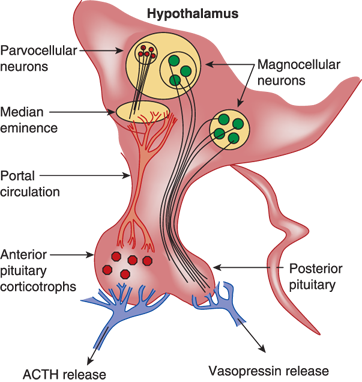
Anatomy of vasopressin-producing neurons. The bilaterally paired paraventricular nucleus (PVN) and the supraoptic nucleus (SON) of the hypothalamus synthesize vasopressin. The PVN contains magnocellular and parvocellular neurons while the SON contains only magnocellular neurons. The magnocellular neurons of the PVN and SON synthesize vasopressin in their soma and carry it through their long axons to the posterior pituitary gland through the pituitary stalk to eventually secrete it from their terminals in the neurohypophysis. Vasopressin released from magnocellular neurons is primarily involved in water balance. The parvocellular neurons that synthesize vasopressin along with corticotrophin-releasing hormone (CRH) have shorter axons that terminate on capillaries of the hypothalamic-hypophyseal portal system in the median eminence of the hypothalamus. Vasopressin secreted here is carried in the portal circulation directly to the anterior pituitary corticotrophs that secrete adrenocorticotropic hormone (ACTH). Vasopressin from the parvocellular neurons, along with CRH, regulates ACTH production and release. The suprachiasmatic nucleus also produces vasopressin that is thought to regulate circadian rhythms (not shown).
Water balance is regulated in two ways: Vasopressin secretion stimulates water reabsorption by the kidney, thereby reducing future water losses, and thirst stimulates water ingestion, which restores past water losses (Figure 8-4). Ideally, these two systems work in parallel to efficiently regulate extracellular fluid tonicity. However, each system by itself can maintain plasma osmolality in the near-normal range. For example, in the absence of vasopressin secretion, but with free access to water, thirst drives water ingestion up to the 5 to 10 L/m2 of urine output seen with vasopressin deficiency. Conversely, an intact vasopressin secretory system can compensate for some degree of disordered thirst regulation. However, when both vasopressin secretion and thirst are compromised, either by disease or iatrogenic means, there is great risk for the occurrence of life-threatening abnormalities in plasma osmolality. In evaluating any disorder of water balance, the clinician should ask whether abnormalities exist in vasopressin secretion, in thirst, or in both systems.
Figure 8-4
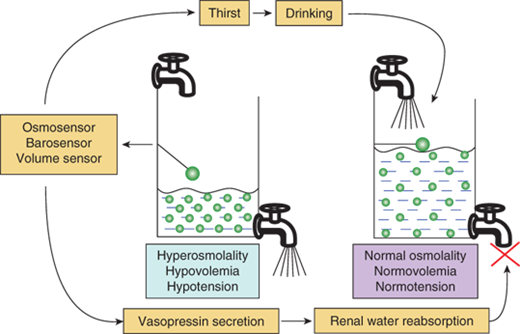
Regulation of water balance. Hyperosmolality, hypovolemia, or hypotension are sensed by osmosensors, volume sensors, or barosensors, respectively, which stimulate the release of vasopressin and the sensation of thirst. Vasopressin acts on the V2 receptor in the kidney to increase water reabsorption and minimize further water loss in the urine. Thirst leads to increased intake of water and the replenishment of the water content of the body. The results of these dual negative feedback loops cause a reduction in hyperosmolality and/or hypotension/hypovolemia.
Vasopressin secretion is stimulated by a rise in the plasma concentrations of osmotically active substances (ie, those that are not freely permeable across cell membranes) principally sodium chloride and glucose (with insulin deficiency). Normal blood osmolality ranges between 280 and 290 mOsm/kg H2O, with the threshold for vasopressin release being approximately 283 mOsm/kg. Above this threshold, an osmosensor located outside the blood-brain barrier near the anterior hypothalamus, which can detect as little as a 1% to 2% change in blood osmolality, signals for posterior pituitary secretion of vasopressin.5 Vasopressin rises in proportion to plasma osmolality, up to a maximum concentration of about 20 pg/mL, when the plasma osmolality is approximately 320 mOsm/kg (Figure 8-5).
Figure 8-5
Vasopressin secretion in response to osmotic and nonosmotic stimuli. Comparison in humans, of the percentage change in osmolality (increase) and pressure or volume (decrease) required to stimulate vasopressin release. While as little as a 1% increase in osmolality results in vasopressin secretion, greater than a 10% to 15% change in volume and pressure is required to stimulate release of vasopressin. However, the magnitude of the vasopressin response to decreases in the volume or pressure is several-fold greater than that in response to changes in osmolality. (Kronenberg HM, Melmed S, Polonsky KS, Larsen PR. William’s Textbook of Endocrinology. 12th ed. Philadelphia, PA: W.B. Saunders; 2011. With permission.)
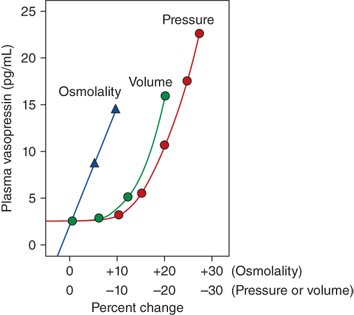
Separate from osmotic regulation, vasopressin is secreted in response to a decrease in intravascular volume or pressure. Afferent baroreceptor pathways arising from the right and left atria and aortic arch (carotid sinus) are stimulated by increasing intravascular volume and stretch of vessel walls, and send signals to the hypothalamic PVN and supraoptic nucleus (SON) to inhibit vasopressin secretion. The pattern of vasopressin secretion in response to volume as opposed to osmotic stimuli is markedly different. While minor changes in plasma osmolality above 280 mOsm/kg evoke linear increases in plasma vasopressin, no change in vasopressin secretion is seen until blood volume decreases by approximately 8%. With intravascular volume deficits exceeding 8%, vasopressin concentration rises exponentially, such that when blood volume and/or pressure decrease by approximately 25%, vasopressin concentrations rise to 20- to 30-fold above normal (see Figure 8-5),6 vastly exceeding those required for maximal antidiuresis, and high enough to cause vasoconstriction via V1 vascular receptors (see later in this chapter).
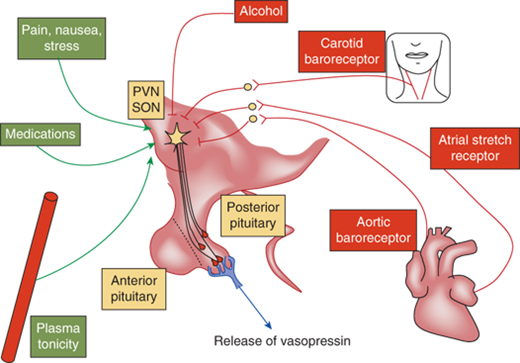
Nausea, pain, hypoglycemia, psychological stress, ethanol, and chlorpropamide are also clinically important triggers for vasopressin release (Figure 8-6). On the other hand, vasopressin secretion is inhibited by glucocorticoids and, because of this, loss of negative regulation of vasopressin secretion occurs in the setting of primary and secondary glucocorticoid insufficiency. Cortisol deficiency both enhances hypothalamic vasopressin production as well as directly impairs free water excretion, important considerations in the evaluation of the patient with hyponatremia, as subsequently discussed.
Figure 8-6
Regulation of vasopressin secretion. Vasopressin is controlled by osmotic and nonosmotic factors. Osmotic regulation occurs through osmosensors in the organum vasculosum of the lamina terminalis (OVLT). An increase in plasma osmolality stimulates secretion of vasopressin. Several nonosmotic factors regulate vasopressin secretion. Blood pressure–controlled regulation of vasopressin is mediated by afferents from baroreceptors in the aorta and the carotid sinus via the cranial nerves IX and X with relays within the brainstem nuclei. Hypovolemia stimulates the secretion of vasopressin via stretch receptors in the atria and pulmonary veins. Nausea is a potent stimulator of vasopressin secretion. While pain, stress, and several medications stimulate the secretion of vasopressin, alcohol inhibits its release. All these factors ultimately act upon the magnocellular vasopressinergic neurons of the PVN and the SON nuclei of the hypothalamus to regulate the synthesis and secretion of vasopressin.
Once in the circulation, vasopressin has a half-life of only 5 to 10 minutes, because of its rapid degradation by a cysteine aminoterminal peptidase, termed vasopressinase. The synthetic analog of vasopressin, DDAVP, is insensitive to aminoterminal degradation, and thus has a much longer half-life of 8 to 24 hours. During pregnancy, the placenta secretes increased amounts of this vasopressinase, resulting in a fourfold increase in the metabolic clearance rate of vasopressin. Normal women compensate with an increase in vasopressin secretion, but women with preexisting deficits in vasopressin secretion or action, or those with increased concentrations of placental vasopressinase associated with liver dysfunction or multiple gestation, may develop diabetes insipidus (DI) in the last trimester, which resolves in the immediate postpartum period. As expected, this form of DI responds to treatment with DDAVP (which is resistant to degradation by vasopressinase), but not with vasopressin.
Vasopressin released from the posterior pituitary and median eminence affects the function of several tissue types by binding to three G-protein–coupled cell surface receptors, designated V1, V2, and V3 (or V1b) (Table 8-1). The major sites of V1 receptor expression are on vascular smooth muscle and hepatocytes, where receptor activation results in vasoconstriction and glycogenolysis, respectively. The concentration of vasopressin needed to significantly increase blood pressure is several-fold higher than that required for maximal antidiuresis. The V3 receptor is present on corticotrophs in the anterior pituitary and acts to increase ACTH secretion.
| Receptor | Target Tissue | Function |
|---|---|---|
| V1 | Vascular smooth muscle, platelets, and hepatocytes | Vasoconstriction, hemostasis, and glycogenolysis |
| V2 | Distal tubule and collecting duct of the kidney | Aquaporin-2–mediated water reabsorption |
| V3 (V1b) | Anterior pituitary corticotrophs | ACTH release |
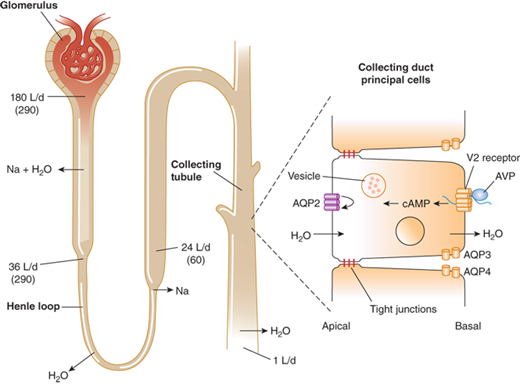
Modulation of water balance occurs through the action of vasopressin upon V2 receptors located primarily on the blood (basolateral) side of cells in the renal collecting tubule. The human V2 receptor gene is located on the long arm of the X chromosome, at the locus associated with congenital, X-linked vasopressin-resistant nephrogenic DI. Vasopressin-induced increases in intracellular cyclic adenosine monophosphate (cAMP) mediated by the V2 receptor trigger a complex pathway of events, resulting in increased permeability of the collecting duct to water and efficient water transit across an otherwise minimally permeable epithelium (Figure 8-7). Activation of a cAMP-dependent protein kinase causes the insertion of aggregates of a water channel, termed aquaporin-2, into the apical (luminal) membrane. Insertion of aquaporin-2 causes up to a 100-fold increase in water permeability of the apical membrane, allowing water movement from the tubule lumen along its osmotic gradient into the hypertonic inner medullary interstitium and excretion of a concentrated urine (see Figure 8-7).7
Figure 8-7
Antidiuretic effect of vasopressin in the regulation of urine volume. In a typical 70-kg adult, the kidney filters approximately 180 L/day of plasma. Of this, approximately 144 L (80%) is reabsorbed iso-osmotically in the proximal tubule and another 8 L (4%-5%) is reabsorbed without solute in the descending limb of Henle loop. The remainder is diluted to an osmolality of approximately 60 mmol/kg by selective reabsorption of sodium and chloride in the ascending limb. In the absence of AVP, the urine issuing from the loop passes largely unmodified through the distal tubules and collecting ducts, resulting in a maximum water diuresis. In the presence of AVP, solute-free water is reabsorbed osmotically through the principal cells of the collecting ducts, resulting in the excretion of a much smaller volume of concentrated urine. This antidiuretic effect is mediated via a G-protein–coupled V2 receptor that increases intracellular cAMP, thereby inducing translocation of aquaporin-2 (AQP-2) water channels into the apical membrane. The resultant increase in permeability permits an influx of water that diffuses out of the cell through AQP-3 and AQP-4 water channels on the basolateral surface. The net rate of flux across the cell is determined by the number of AQP-2 water channels in the apical membrane and the strength of the osmotic gradient between tubular fluid and the renal medulla. Tight junctions on the lateral surface of the cells serve to prevent unregulated water flow. (Reproduced with permission from Longo DL, Fauci AS, Kasper DL: Harrison’s Principles of Internal Medicine, 18th edition. McGraw-Hill Companies, 2012.)
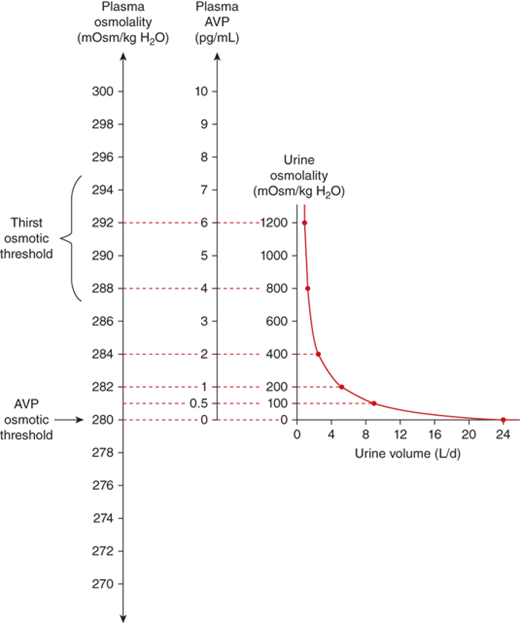
The sensation of thirst is determined by hypothalamic neurons anatomically distinct from those that make vasopressin. It makes physiologic sense that the threshold for thirst (∼293 mOsm/kg) is slightly higher than that for vasopressin release (Figure 8-8). Otherwise, during the development of hyperosmolality, the initial activation of thirst and water ingestion would result in polyuria without activation of vasopressin release, causing a persistent diuretic state.8
Figure 8-8
Physiological relations between plasma osmolality, plasma AVP concentrations, urine volume, and urine osmolality in healthy humans.61 Note that the osmolality at which the secretion of vasopressin is stimulated is below that at which thirst is stimulated. (Adapted from Robinson AG: Disorders of antidiuretic hormone secretion, Clin Endocrinol Metab Feb;14(1):55-88, 1985, with permission.)
Hypernatremia along with hyponatremia constitutes the commonest electrolyte abnormality in children.
Hypernatremia usually occurs when free water loss exceeds salt loss from the body.
Hypernatremia is defined as a serum sodium concentration greater than 145 mmol/L. It occurs most often in the setting of dehydration, due to either pure water loss or water loss that exceeds salt loss. Hypernatremia can occur due to either free water loss or excessive intake of sodium. The latter usually causes transient hypernatremia. In the presence of an intact thirst mechanism, the ability to drink water, and intact renal function, it is followed by water intake leading to volume expansion and natriuresis, thus normalizing the serum sodium levels. Similarly, excessive loss of free water is usually compensated by increased intake driven by the intact thirst mechanism. Inability to drink water freely as seen in infants and neurologically impaired children can thus lead to hypernatremia from either excessive ingestion of sodium or from excessive free water loss. Infants in particular also have a limited ability to excrete the osmotic solute load from their kidneys and hence can have persistent hypernatremia from accidental or deliberate inclusion of excessive amounts of salt in their food.
Children with significant hypernatremia progress from irritability to lethargy and coma. Neurologic signs such as nuchal rigidity, hypertonia, and hyperreflexia may be present. Seizures and rhabdomyolysis can occur. Shrinkage of cells in the brain in acute hypernatremia can lead to the rupture of bridging veins causing intracranial hemorrhage. Venous infarcts from venous sinus thrombosis can also be seen. A mortality rate of 15% has been reported in children with hypernatremia.9
A diagnostic algorithm for hypernatremia (Figure 8-9) in most children with a normal thirst mechanism and a normal ability to drink water is thus most often limited to the identification of the source of the free water loss. The presence of polyuria with dilute urine confirms renal loss of free water. The diagnostic approach for polyuria will be discussed subsequently in this chapter. Free water may be lost relatively in excess to sodium loss through nonrenal routes such as the skin as in burns and excessive sweating, the respiratory tract or the gastrointestinal tract as occurs with diarrhea, or through fistulae. Because hypernatremia causes intracellular dehydration due to osmotic shift of intracellular fluid to the extracellular compartment, the extent of dehydration is often underestimated during physical examination. In addition to other signs of dehydration, the skin has been classically described as having a “doughy” consistency (due to increased turgor) in hypernatremic dehydration. Laboratory investigations should include serum sodium, osmolality, blood urea nitrogen (BUN), and creatinine, along with urine sodium and osmolality.
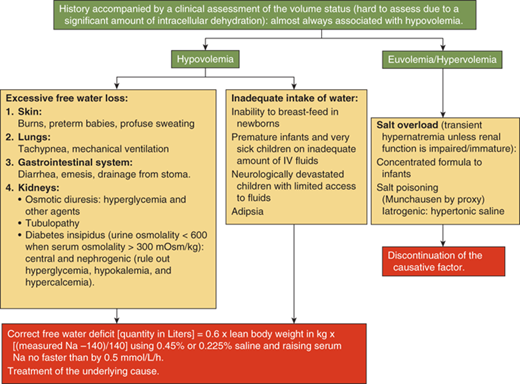
The goals of treating hypernatremia should be to correct the hypovolemia and to normalize the serum sodium level. The management of hypernatremia should involve identification of the underlying cause and the duration of the hypernatremia. Treatment is directed at the underlying cause when one can be identified. An estimation of the free water deficit is made using the formula:
This free water deficit is best corrected slowly with oral or nasogastric free water over a period of 24 to 48 hours. Ongoing losses should be replaced simultaneously while closely monitoring the clinical status and the serum electrolytes. In children who are unable to receive free water through the enteral route, a parenteral solution containing either 5% dextrose (D5), or 0.45% to 0.225% saline can be used.
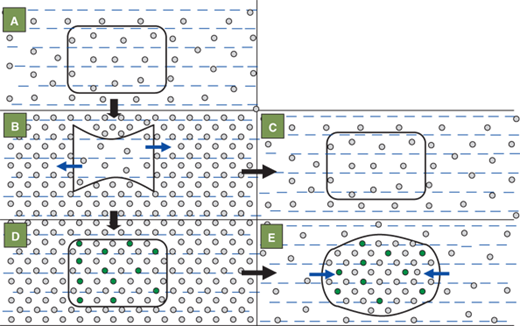
The brain cells adapt to hypernatremia over a period of days, by increasing the intracellular osmolyte concentration in order to prevent intracellular dehydration and cell shrinkage (Figure 8-10). They do so by increasing the levels of electrolytes, amino acids such as taurine, and glutamine and other osmotically active substances such as myoinositol, and betaine.10 While this process is protective during chronic hypernatremia, rapid lowering of the serum sodium can cause swelling of the brain cells. The brain being enclosed within the cranial vault, which is nonexpandable after the closure of the fontanelles, can herniate infratentorially. Seizures, coma, and even death can occur with rapid lowering of chronically elevated serum sodium levels. Hence it is widely accepted that the serum sodium level should be corrected at least over a period of 48 hours. It should not be lowered more rapidly than a rate of 15 mmol/L/day.
Figure 8-10
The effects of hypernatremia and its rapid correction on intracellular osmolality and size of the brain cell. The shape and size of cell are normal in the eunatremic state (A) in which the osmolalities within and outside the cell are equal. Acute hypernatremia (B) causes an osmotic fluid shift out of the cell (blue arrows) leading to cell shrinkage. Rapid correction of the acute extracellular hypernatremia (C) once again equalizes the osmolality within and outside the cell and thus restores the cell shape and size. If hypernatremia persists uncorrected beyond 24 hours (D), the cell tries to regain its original shape and size by generating new osmolytes (green circles) to equilibrate its osmolality with that of the outside. Rapidly lowering the serum sodium and extracellular osmolality at this stage (E) causes cell swelling due to osmotic flow of water into the cell (blue arrows). The cell is unable to eliminate some of its osmolytes rapidly enough to once more equilibrate the intra- and extracellular osmolalities.
Central (hypothalamic, neurogenic, or vasopressin-sensitive) DI in children is most often the result of surgical or accidental trauma to vasopressin neurons, congenital anatomical hypothalamic or pituitary defects, or neoplasms. It can also be caused by infiltrative, autoimmune, and infectious diseases affecting vasopressin neurons or fiber tracts, and, least commonly, by disorders of vasopressin gene structure (Table 8-2). In different case series published in the literature, an underlying cause could not be identified in between 9% and 55% of children and young adults affected by central DI.11,12
| Congenital: |
| Septo-optic dysplasia/optic nerve hypoplasia |
| Ectopia/hypogenesis of the posterior pituitary |
| Holoprosencephaly |
| Other midline craniofacial defects |
| Familial: |
| Autosomal dominant |
| Autosomal recessive |
| Wolfram syndrome (DIDMOAD)—autosomal recessive |
| Acquired: |
| Postoperative: with or without triple-phase response |
| Traumatic: transection of the hypothalamic-pituitary stalk |
| Septic shock (infarction) |
| Hypoxic brain injury |
| Neoplasms: |
| Germinoma with or without a pinealoma |
| Craniopharyngioma |
| Metastatic tumors: leukemias |
| Optic glioma |
| Infiltrative: |
| Langerhans cell histiocytosis |
| Sarcoidosis |
| Pulmonary granulomatous diseases including granulomatosis with angiitis |
| Autoimmune: lymphocytic hypophysitis |
| Drugs: ethanol, phenytoin, opiate antagonists, and α-adrenergic agonists |
| Infectious: (meningitis/encephalitis) |
| Cryptococcus neoformans and Listeria monocytogenes |
| Mycobacterium tuberculosis, toxoplasmosis, and congenital CMV |
| Brain aneurysms and Rathke cleft cysts |
| Pregnancy: placental vasopressinase |
| Sheehan syndrome (postpartum hemorrhage) |
| Idiopathic |
The most common cause of central DI is the neurosurgical destruction of vasopressin neurons following pituitary-hypothalamic surgery. It is important to distinguish polyuria associated with the onset of acute postsurgical central DI from polyuria due to the normal diuresis of fluids given during surgery. In both cases, the urine may be very dilute and of high volume, exceeding 200 mL/m2/h. However, in the former case, serum osmolality will be high, whereas in the latter case it will be normal. A careful examination of the intraoperative record should also help distinguish between these two possibilities. The axons of vasopressin-containing magnocellular neurons extend uninterrupted to the posterior pituitary over a distance of approximately 10 mm. These axons terminate at various levels within the stalk and gland (see Figure 3-3). Because surgical interruption of these axons can result in retrograde degeneration of hypothalamic neurons, lesions closer to the hypothalamus will affect more neurons and cause greater permanent loss of hormone secretion. Not infrequently, a “triple-phase” response is seen (Table 8-3). Following surgery, an initial phase of transient DI is observed, lasting 1.5 to 2 days, which is possibly due to edema in the area interfering with normal vasopressin secretion. If significant vasopressin cell destruction has occurred, this is often followed by a second phase of syndrome of inappropriate antidiuretic hormone (SIADH), which may last up to 10 days, and is due to the unregulated release of vasopressin, by dying neurons. A third phase of permanent DI may follow if more than 90% of vasopressin cells are destroyed. Usually, a marked degree of SIADH in the second phase portends significant permanent DI in the final phase of this response. In a small study, the “triple-phase response” was observed in one out of three children who underwent surgery for a craniopharyngioma.13 In patients with coexisting vasopressin and cortisol deficits (eg, in combined anterior and posterior hypopituitarism following neurosurgical treatment of craniopharyngioma), symptoms of DI may be masked because cortisol deficiency impairs renal free water clearance, as discussed subsequently. In such cases, institution of glucocorticoid therapy alone may precipitate polyuria, leading to the diagnosis of DI.
| Phase | Condition | Underlying Cause | Duration |
|---|---|---|---|
| First | Transient diabetes insipidus | Impaired secretion: local edema | 0.5-2 days |
| Second | SIADH | Release of vasopressin from dying neurons | 1-10 days |
| Third | Permanent diabetes insipidus | Cell loss | Permanent |
Trauma to the base of the brain can cause swelling around or severance of these axons, resulting in either transient or permanent DI. Permanent DI can occur after seemingly minor trauma. Approximately one-half of patients with fractures of the sella turcica will develop permanent DI, which may be delayed by up to 1 month following the trauma, during which time neurons of severed axons may undergo retrograde degeneration. Septic shock and postpartum hemorrhage associated with pituitary infarction (Sheehan syndrome) may involve the posterior pituitary with varying degrees of DI. DI is never associated with cranial irradiation of the hypothalamic-pituitary region.
Midline brain anatomical abnormalities such as septo-optic dysplasia (SOD)/optic nerve hypoplasia (ONH) with agenesis of the corpus callosum, holoprosencephaly, and familial pituitary hypoplasia with absent stalk may be associated with central DI, which usually presents in the first month of life, often with signs of anterior pituitary dysfunction, such as jaundice, and, in boys, microphallus. Central DI due to midline brain abnormalities may be accompanied by defects in thirst perception.
Because hypothalamic vasopressin neurons are distributed over a large area within the hypothalamus, tumors that cause DI must either be very large, be infiltrative, or be strategically located at the point of convergence of the hypothalamic-neurohypophyseal axonal tract in the infundibulum. Germinomas and pinealomas typically arise near the base of the hypothalamus where vasopressin axons converge prior to their entry into the posterior pituitary, and for this reason are among the most common primary brain tumors associated with DI. Germinomas causing the disease can be very small and undetectable by magnetic resonance imaging (MRI) for several years following the onset of polyuria. For this reason, serial quantitative measurements of the β-subunit of human chorionic gonadotropin (β-hCG), often secreted by germinomas and pinealomas, and regularly repeated MRI scans should be performed in children with idiopathic or unexplained DI. Long-term surveillance can identify, sometimes after as long as 21 years, an underlying cause that was not apparent at the time of initial diagnosis.14
Empty sella syndrome, possibly due to unrecognized pituitary infarction, can be associated with DI in children. Craniopharyngiomas and optic gliomas, when very large, can also cause central DI, although this is more often a postoperative complication of the treatment for these tumors. Hematologic malignancies can cause DI. In some cases, such as with acute myelocytic leukemia, the cause is infiltration of the pituitary stalk and sella.
Langerhans cell histiocytosis and lymphocytic hypophysitis are the most common types of infiltrative disorders causing central DI. Approximately 10% of patients with histiocytosis will have DI. These patients tend to have more serious, multisystem disease for longer periods of time than those without DI, and anterior pituitary deficits often accompany posterior pituitary deficiency. MRI characteristically shows thickening of the pituitary stalk. Radiation treatment to the pituitary region within 14 days of onset of symptoms of DI may result in return of vasopressin function. In adults, lymphocytic infundibuloneurohypophysitis may account for over one-half of patients with “idiopathic” central DI. It is a rare cause of DI in children. This entity may be associated with other autoimmune diseases. Image analysis discloses an enlarged pituitary and thickened stalk, and biopsy of the posterior pituitary reveals lymphocytic infiltration of the gland, stalk, and magnocellular hypothalamic nuclei. A necrotizing form of this entity has been described, which also causes anterior pituitary failure and responds to glucocorticoid treatment. DI can also be associated with pulmonary granulomatous diseases including sarcoidosis and granulomatosis with angiitis.
Infections involving the base of the brain, such as meningococcal, cryptococcal, Listeria, and toxoplasmosis meningitis, congenital cytomegalovirus infection, and nonspecific inflammatory disease of the brain can cause central DI. The disease is often transient, suggesting that it is due to inflammation rather than destruction of vasopressin-containing neurons.
Familial, autosomal dominant central DI is manifest within the first half of the first decade of life. Vasopressin secretion, initially normal, gradually declines until DI of variable severity ensues. Patients respond well to vasopressin-replacement therapy. The disease has a high degree of penetrance, but it may be of variable severity within a family. Several different single nucleotide mutations in the vasopressin structural gene have been found to cause the disease, with most occurring in the neurophysin region. Endoplasmic reticulum stress from misfolding of the aberrant protein has been hypothesized to be the possible cause for the premature loss of the magnocellular vasopressin neurons seen in this disease.15 Vasopressin deficiency is also found in the DIDMOAD syndrome, consisting of Diabetes Insipidus, Diabetes Mellitus, Optic Atrophy, and Deafness. This syndrome complex, also known as Wolfram syndrome, is caused by recessive mutations in the WFS1 gene located on chromosome 4p16. This gene codes for wolframin, a transmembrane protein localizing to the endoplasmic reticulum that is believed to either act as a calcium channel or as a regulator of a calcium channel.16
Whereas normal children have a nocturnal rise in plasma vasopressin associated with an increase in urine osmolality and a decrease in urine volume, those with primary enuresis have a blunted or absent rise in vasopressin, and excrete a higher urine volume of lower tonicity. This has suggested that enuretic children have a primary deficiency in vasopressin secretion, although the same outcome could be caused solely by excessive water intake in these children. The use of the V2 agonist, DDAVP, is highly effective in abolishing bed-wetting episodes, although relapse is high once therapy is stopped. This therapy must be used with caution, as fluid intake must be limited while a child is exposed to the antidiuretic action of DDAVP to guard against water intoxication.
Patients with otherwise untreated DI crave cold fluids, especially water. With complete central DI, maximum urine concentrating ability is approximately 200 mOsm/kg. An average 10-year-old child with a body surface area of 1 m2 would then require a urine output of 2.5 L of urine to excrete an average daily solute load of 500 mOsm/m2. Insensible (nonrenal) fluid losses in this example would amount to 500 mL/day (500 mL/m2/day). In order to maintain normal plasma tonicity, this child’s fluid intake must match his/her fluid loss of 3 L/day. With an intact thirst mechanism and free access to oral fluids, a person with complete DI can maintain plasma osmolality and sodium in the high-normal range, although at great inconvenience. Furthermore, long-standing intake of these volumes of fluid in children can lead to hydroureter, and even hyperfluorosis in communities that provide fluoridated water.
There are two situations in which central DI is sometimes treated solely with high levels of fluid intake, without vasopressin. Vasopressin therapy coupled with excessive fluid intake (usually > 1 L/m2/day as discussed subsequently) can result in unwanted hyponatremia. Because neonates and young infants receive all of their nutrition in liquid form, the obligatory high oral fluid requirements for this age (3 L/m2/day), combined with vasopressin treatment, are likely to lead to this dangerous complication.17 Thus, neonates may be better managed with fluid therapy alone. The use of breast milk or Similac 60/40 formula reduces the renal solute load by 20% to 30%. This in turn reduces the urine volume in infants with DI by 20% to 30%. Chlorothiazide (oral dose 5 mg/kg twice daily) is a diuretic that increases the urine osmolality and decreases the urine output through mechanisms that are unclear. The latter two therapeutic options significantly reduce the amount of free water supplementation needed in infants with both forms of DI. However, supplementation with 20 to 30 mL of free water for every 120 to 160 mL of formula might still be required.18 Infants with central DI have also been successfully treated with subcutaneous injections of DDAVP (initial dose 0.002-0.1 μg/kg once daily, titrated upward and given twice daily if needed) until they are eating solid food. Therapy in infants with subcutaneous injections of DDAVP has been associated with far fewer episodes of hyponatremia and hypernatremia than with either intranasal or oral DDAVP.19
In the acute postoperative management of central DI occurring after neurosurgery in young children, vasopressin therapy may be successfully used, but extreme caution must be exerted with its use. While under the full antidiuretic effect of vasopressin, a patient will have a urine osmolality of approximately 1000 mOsm/kg and become hyponatremic if she/he receives an excessive amount of fluids, depending on the solute load and nonrenal water losses. With a solute excretion of 500 mOsm/m2/day, normal renal function, and nonrenal fluid losses of 500 mL/m2/day, fluid intake of greater than 1 L/m2/day will result in hyponatremia. In addition, vasopressin therapy will mask the emergence of the SIADH phase of the “triple-phase” neurohypophyseal response to neurosurgical injury (as discussed earlier). For these reasons, it is often best to manage acute postoperative DI in young children with fluids alone, avoiding the use of vasopressin. This method consists of matching input and output hourly using between 1 and 3 L/m2/day (40-120 mL/m2/h). If intravenous therapy is used, a basal 40 mL/m2/h should be given as D5 in 1/4 normal saline (normal saline = 0.9% sodium chloride) and the remainder, depending on the urine output, as 5% dextrose inwater (D5W). Potassium chloride (40 mmol/L) may be added if oral intake is to be delayed for several days. No additional fluid should be administered for hourly urine volumes under 40 mL/m2/h. For hourly urine volumes above 40 mL/m2/h, the additional volume should be replaced with D5W up to a total maximum of 120 mL/m2/h. For example, in a child with a surface area of 1 m2 (∼30 kg), the basal infusion rate would be 40 mL/h of D5 in 1/4 normal saline. For an hourly urine output of 60 mL, an additional 20 mL/h D5W would be given, for a total infusion rate of 60 mL/h. For urine outputs above 120 mL/h, the total infusion rate would be 120 mL/h. In the presence of DI, this will result in a serum sodium in the 150 mmol/L range and a mildly volume contracted state, which will allow one to assess both thirst sensation as well as the return of normal vasopressin function or the emergence of SIADH. Patients may become mildly hyperglycemic with this regimen, particularly if they are also receiving postoperative glucocorticoids. However, this fluid management protocol, because it does not use vasopressin, prevents any chance of hyponatremia.
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


