Plasma Cell Myeloma and Plasmacytoma
 EPIDEMIOLOGY AND ETIOLOGY
EPIDEMIOLOGY AND ETIOLOGY
Plasma cell neoplasms account for 22% of all mature B-cell neoplasms in the Surveillance, Epidemiology, and End Results (SEER) program of the United States.1 The majority of plasma cell neoplasms are multiple myeloma, with solitary plasmacytoma accounting for ≤6% of cases, and plasma cell leukemia rarely. Although the incidence of multiple myeloma gradually increased in the 1970s through 1990s, recently there has been a plateau in the U.S. incidence from 1992 to 2008.2 Data from SEER indicate an incidence in the United States of 7.2 per 100,000 men per year and 4.6 per 100,000 women during the period 2004 to 2008,2 and for 2011, it is estimated that there will be 20,520 new cases and 10,610 deaths due to multiple myeloma in the United States.3 The incidence exceeds that of Hodgkin lymphoma and is about one-quarter that of non-Hodgkin lymphoma. The incidence rises with advancing age, with a median age at diagnosis of 70 years,4 and <1% of cases are diagnosed in those younger than 35. Nonregistry studies usually report a lower median age ranging from 60 to 66 years.5,6 There is a slight male predominance, and for black Americans, the incidence and mortality rates are approximately double that for whites. The 5-year relative survival rates have increased, from 26% in 1975 to 40% in 2003.2
Little is known about the cause of multiple myeloma. There are studies reporting association with prior exposure to radiation (e.g., atomic bomb survivors in Hiroshima)7 and certain chemicals such as petroleum products.8,9 It is now thought that all cases of myeloma are preceded by monoclonal gammopathy of unknown significance (MGUS).10,11,12
TABLE 81.1 THE SPECTRUM OF MYELOMA

 PATHOPHYSIOLOGY
PATHOPHYSIOLOGY
Multiple myeloma arises from malignant transformation of a late-state B cell. Although the full cascade of genetic abnormalities has yet to be defined, one of the earliest genetic events is the illegitimate switch recombination of partner oncogenes into the immunoglobulin heavy chain (IgH).13 Other events may occur such as cytogenetic hyperploidy and up-regulation of cell cycle control genes. The result of these genetic abnormalities is the development and propagation of a clonal population of B cells within the bone marrow; this, however, is common and can be seen in up to 5% of the general population over the age of 70 (MGUS).14 Most of these will not go on to develop myeloma, so there must be additional events to create the malignant phenotype of multiple myeloma. These secondary events may include mutations of kinases, deletions of chromosomes, and up-regulation of enzymes such as c-myc.15 Having sustained a secondary event, the malignant plasma cells begin to proliferate in the bone marrow microenvironment, producing monoclonal proteins and causing osteolytic bone disease. The slow accumulation of these malignant cells gradually results in the characteristic clinical features of myeloma: anemia, bone resorption, hypercalcemia, renal failure, and immunodeficiency. Established myeloma is sustained by a number of microenvironment features, including the bone marrow stroma itself and the cytokines interleukin-6 and insulin-like growth factor-1.16 The bone disease that arises in myeloma appears to be mediated in part by Rank ligand/osteprotegrin and the Wnt signaling antagonist Dickkopf1.17
TABLE 81.2 DIAGNOSTIC WORKUP FOR PLASMA CELL NEOPLASMS

 CLINICAL PRESENTATION
CLINICAL PRESENTATION
Multiple myeloma has a wide clinical spectrum, ranging from the preclinical condition of MGUS to the most aggressive form, plasma cell leukemia (Table 81.1). In all cases, a plasma cell clone exists, and the secretion of a monoclonal protein by these plasma cells, along with their interaction with the bone marrow environment, is the source of organ damage in patients with this illness.16 These concepts have become particularly important as the molecular mechanisms by which the disease progresses through these “stages” provide essential information that may help us to better understand the disease and its potential therapies.
Monoclonal Gammopathy of Unknown Significance
MGUS has traditionally been considered a benign or a premalignant condition, in which only a small proportion of patients will progress to multiple myeloma or related diseases (Table 81.1). In MGUS, the monoclonal protein is <3 g/dL and the bone marrow clonal plasma cells are <10% with no related organ damage. This condition is likely much more common than initially thought, as it has been documented in 3% of the population and 5% in those over the age of 70.14 The risk of transformation to myeloma and related diseases (such as amyloidosis or Waldenstrom’s macroglobulinemia) has been estimated at 1% per year, based on a 30-year follow-up of 1,384 patients at the Mayo Clinic.10
Asymptomatic Multiple Myeloma (Smoldering Myeloma)
This category of myeloma represents an intermediate form of myeloma whereby patients do meet serological monoclonal protein and bone marrow criteria for the diagnosis of myeloma (in excess of 10% clonal plasmacytosis) but have yet to develop evidence of end organ damage (Table 81.1). These patients are not significantly anemic, do not have renal insufficiency, and do not have bony disease. Although the risk of transformation to multiple myeloma is much higher than in MGUS (20% per year), some patients’ disease may remain asymptomatic without significant progression for many years. These patients generally do not require therapy but should be followed closely to monitor for progression.
Solitary Plasmacytomas
The median age at diagnosis of solitary plasmacytoma (SP) is 55 to 65 years, on average about 10 years younger than patients with multiple myeloma.18,19–20 Males are affected predominately (male-to-female ratio 2:1).18 A diagnosis of SP is made if all the following criteria are satisfied at presentation: a histologically confirmed single lesion with negative skeletal imaging outside the primary site, normal bone marrow biopsy (<10% monoclonal plasma cells), and no myeloma-related organ dysfunction.21 A monoclonal protein is present in 30% to 75% of cases (particularly for an osseous presentation), and the level is usually minimally elevated (IgG <3.5 g/dL, IgA <2.0 g/dL, and urine monoclonal κ or λ <1.0 g/24 hours).21,22
The disease more commonly presents in bone (80%). Such cases are considered stage I multiple myeloma according to the Durie Salmon staging system.23 The most common location is the vertebra.18 Patients with bone involvement often present with pain, neurologic compromise, and occasionally pathologic fracture. A lytic lesion is typical, with or without adjacent soft tissue mass. Less commonly, SP presents in an extramedullary site (20%), usually as a mass in the upper aerorespiratory passages, and produces local compressive symptoms.18,19,24,25 The histologic diagnosis of extramedullary plasmacytoma (EMP) can be difficult, with the main differential diagnosis being extranodal marginal zone lymphoma (mucosa-associated lymphoid tissue type), where there can be extensive infiltration by plasmacytoid cells.24,26
Multiple Myeloma
By definition, myeloma involves end organ damage, described by the mnemonic CRAB (Calcium elevation, Renal insufficiency, Anemia, and Bone disease).27 Bone pain and symptoms due to anemia, such as easy fatigability, are the most common.5 Because of the myriad effects of the disease, other insidious symptoms can result from a combination of hypercalcemia, renal impairment, infection, neurologic compression, and occasionally, hyperviscosity. Bone disease manifesting as generalized osteopenia and multiple lytic bone lesions can frequently lead to pathologic fractures. In the vertebral column, this often results in a diminished height. Sclerotic lesions at presentation are rare.
Laboratory evaluation generally confirms anemia, high erythrocyte sedimentation rate, and a variable degree of granulocytopenia and thrombocytopenia. An abnormal monoclonal immunoglobulin (M protein) in the blood or urine is characteristic,5 most commonly IgG or IgA. Biclonal disease is also recognized, and rarely, nonsecretory disease. In up to 10% of cases, only monoclonal light chains are detected. It is important to assess for hypercalcemia, renal dysfunction, and integrity of the skeleton because these complications require appropriate management. A constellation of polyneuropathy, organomegaly, endocrinopathy, M protein, and skin changes characterize a rare plasma cell dyscrasia known as POEMS syndrome.28
Plasma Cell Leukemia
This is a very rare variant of multiple myeloma, where the proliferation of plasma cells is not confined to the bone marrow but may be detected in the peripheral blood. It carries a very poor prognosis, with median survival <1 year and shortest when occurring as secondary plasma cell leukemia.29,30 There is currently no standard therapy for this condition, but patients are usually treated with high-dose, multiagent chemotherapeutic regimens or with experimental therapies.
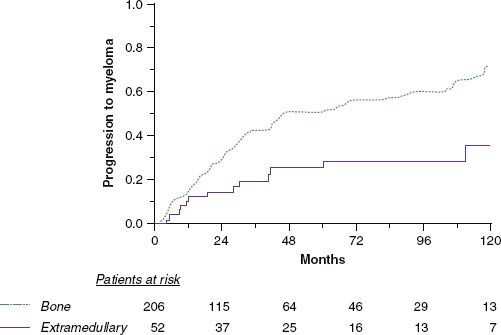
TABLE 81.3 STAGING OF MULTIPLE MYELOMA (DURIE SALMON AND THE NEW INTERNATIONAL STAGING SYSTEM)
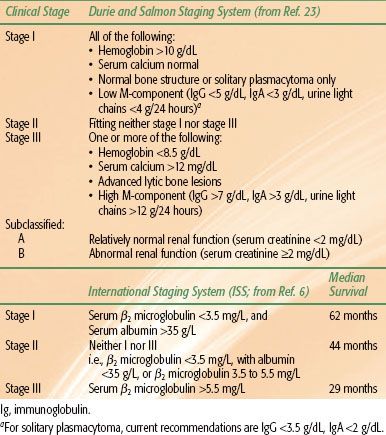
FIGURE 81.1. Probability of progression to multiple myeloma according to bone (dotted line) versus extramedullary (solid line) solitary plasmacytoma in 258 patients (P = .0009). (From Ozsahin M, Tsang RW, Poortmans P, et al. Outcomes and patterns of failure in solitary plasmacytoma: a multicenter Rare Cancer Network study of 258 patients. Int J Radiat Oncol Biol Phys 2006;64(1):210–217, copyright 2006, with permission from Elsevier.)
 DIAGNOSTIC WORKUP AND STAGING
DIAGNOSTIC WORKUP AND STAGING
The recommended tests for the diagnosis of plasma cell neoplasms are outlined in Table 81.2. The most important components relate to the measurement and quantification of the M protein, bone marrow examination with ancillary studies, serum β2 microglobulin and albumin, and diagnostic imaging. The M protein should be measured with serum protein electrophoresis. Quantification of the monoclonal Ig with immunofixation techniques is also acceptable and especially useful if the M component is at a low level. If no M protein is detectable, assays for free light chains should be performed in the serum and in the urine (Bence-Jones proteinuria). The standard imaging is the skeletal survey, as radionuclide bone scan usually does not detect lytic disease and has limited value.21 For localized areas of concern, both computed tomography (CT) or magnetic resonance imaging (MRI) should be liberally utilized. MRI is preferred to assess the extent of vertebral disease and the presence of spinal cord or nerve root compression. With advances in diagnostic imaging, it is likely that “stage migration” has occurred.31 It has been documented that some patients with presumed solitary plasmacytoma of bone will be upstaged following the detection of multiple vertebral lesions or bone marrow disease by MRI or by 18-fluorine (18F) fluorodeoxyglucose positron emission tomography (FDG-PET).32,33–34 The optimal role of PET in myeloma is yet to be determined but will likely evolve rapidly, and it will likely be of most benefit in nonsecretory disease.35,36–37 The staging criteria for the historical Durie Salmon staging system are detailed in Table 81.3.23 The newer International Myeloma Staging System (ISS) is simple, validated, and of importance particularly for present and future clinical trials (Table 81.3).6 Criteria for the diagnosis of MGUS and asymptomatic (smoldering) myeloma are also well established.21,38
 PROGNOSTIC FACTORS
PROGNOSTIC FACTORS
Solitary Plasmacytoma
With respect to local control, tumor bulk appears to be an important unfavorable factor. Tumors <5 cm achieved a high level of local control with 35 Gy, whereas those ≥5 cm had a local failure rate of 58% (7 of 12 patients, total dose range 25 to 50 Gy).20 The importance of tumor bulk is also supported by other studies.18,39,40
Age is a factor that affects the risk of progression to myeloma in some series20,41,42–43 but not in others.18,39,40,44,45 A bony presentation has been consistently demonstrated to have a significantly higher risk of subsequent development of myeloma with a 10-year rate of 76%, compared with an extramedullary presentation where the 10-year rate was 36% (Fig. 81.1).18 Subclinical bone disease, either detected as generalized osteopenia46 or abnormal MRI scan of the spine,33,34,47 predicts for rapid progression to symptomatic multiple myeloma. A suppression of the normal immunoglobulin classes has been shown to correlate with a higher risk of progressing to myeloma.46,48 Where there was an elevation of M protein pretreatment, persistence of the M protein following radiation therapy (RT) predicts for progression to myeloma.22,32 Many of these factors reflect the presence of occult myeloma. Therefore, it is not surprising that generalized disease becomes manifest once the local disease is controlled. Pathologic factors have been examined in some studies, with the finding that anaplastic plasmacytomas (those with a higher histologic grade)49 and those tumors expressing a high level of angiogenesis are associated with a poor outcome.50 Anaplastic plasmacytomas share some common pathologic and clinical features with aggressive B-cell lymphomas (plasmablastic type) and can arise in the context of immunosuppression and Epstein-Barr virus infection.51,52
Multiple Myeloma
Analysis of over 1,000 patients evaluated at the Mayo Clinic revealed the following adverse prognostic risk factors: Eastern Cooperative Oncology Group performance status 3 or 4, serum albumin <3 g/dL, serum creatinine ≥2 mg/dL, platelet count <150,000/μL, age ≥70 years, β2 microglobulin >4 mg/L, plasma cell labeling index ≥1%, serum calcium ≥11 mg/dL, hemoglobin <10 g/dL, and bone marrow plasma cell percentage ≥50%.5 The ISS has been validated to assist in prognostication.6 Over 10,000 patients were evaluated, and the three-stage system was developed based on two variables: serum albumin and β2 microglobulin (Table 81.3). In addition to stage, cytogenetic abnormalities affect prognosis. Some abnormalities demonstrated to carry a poorer prognosis include: deletion of chromosome 13,53 presence of the t(4;14) translocation,54 and p53 deletion.55 Risk stratification by means of conventional cytogenetics and fluorescence in situ hybridization not only influences prognosis but now also affects therapeutic choices.56
 MANAGEMENT OF SOLITARY PLASMACYTOMA
MANAGEMENT OF SOLITARY PLASMACYTOMA
RT is the standard treatment for solitary plasmacytoma. Surgery should be considered for structural instability of bone or rapidly progressive neurologic compromise such as spinal cord compression.21,57,58 For patients treated with gross tumor excision, RT is still indicated due to a high likelihood of microscopic residual disease. Surgery alone without RT leads to an unacceptably high local recurrence rate.18 A review of the literature for solitary bone plasmacytoma (Table 81.4) indicates a high local control rate with RT (79% to 95%), yet a modest overall survival of approximately 50% at 10 years. This is due to a high rate of progression to multiple myeloma in the bone plasmacytomas, a finding consistently reported from all series.18,20,22,40–41,42,46,48,59,60 As shown in Table 81.4, over 60% of patients with solitary bone tumor progressed to myeloma, at a median of 2 to 3 years after treatment. When actuarial methods were not used, the progression rate is slightly lower (crude rates ranges, 53% to 54%).40,59 Therefore, solitary plasmacytoma of bone appears to be an early form of multiple myeloma. Studies have documented about 29% to 50% of patients with apparent solitary plasmacytoma will have multiple asymptomatic lesions detected in the spine on MRI.33,34,61 Provided that all the other diagnostic criteria for solitary plasmacytoma are satisfied, it is still appropriate to treat with local RT to the presenting site.57 For these patients the risk of developing symptomatic myeloma in a short time is high.33,34,47,62 Chemotherapy can be started at the time of symptomatic progression. The presence of low level M protein preradiation is extremely common and is not associated with a higher risk of progression to multiple myeloma. However, its persistence following radiation is highly predictive of subsequent systemic failure,22,32,46,63 attesting to the importance of monitoring this as part of posttreatment follow-up. It has been observed that some patients recur with plasmacytoma(s) of bone or soft tissues, without bone marrow involvement.40,41,64 This is infrequent and the subsequent development of multiple myeloma is high, 75% in one series.41
The addition of adjuvant chemotherapy is theoretically attractive, both in enhancing local control and eradicating subclinical disease to prevent the development of myeloma. One randomized trial suggested a benefit with adjuvant melphalan and prednisone given for 3 years after RT.65 With a median follow-up of 8.9 years, those treated with chemotherapy had a myeloma progression rate of 12%, whereas with RT alone it was 54%. However, this was a small study, and the risk of diminishing stem cell reserve or inducing leukemia makes the prolonged use of alkylating agents an undesirable treatment option for most patients.
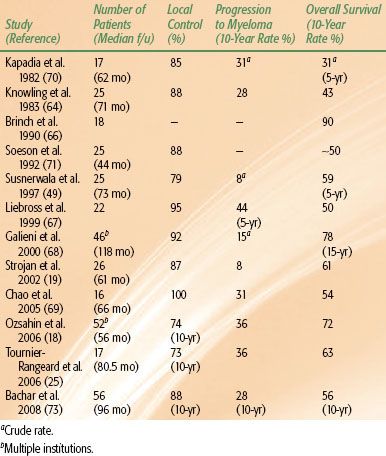
In the management of EMP, while complete surgical excision may be curative for small lesions, most patients with larger lesions or with tumor location not amenable to complete excision should receive local RT. Postoperative RT is indicated for incompletely excised lesions. In contrast to bone plasmacytoma, EMPs are frequently controlled with local radiation (Table 81.5), with a lower rate of progression to myeloma, ranging from 8% to 44%,18,19,25,49,64,66,67–68,69–73 indicating a significant proportion of patients are cured of their disease. Although the 10-year survival varies widely in the reported literature (range, 31% to 90%), the two largest series report 10-year survival rates of 72%18 and 78%.68 The issue of dose will be discussed later.
TABLE 81.4 SOLITARY PLASMACYTOMA OF BONE: REPRESENTATIVE TREATMENT RESULTS (SERIES INCLUDING MORE THAN 30 PATIENTS)
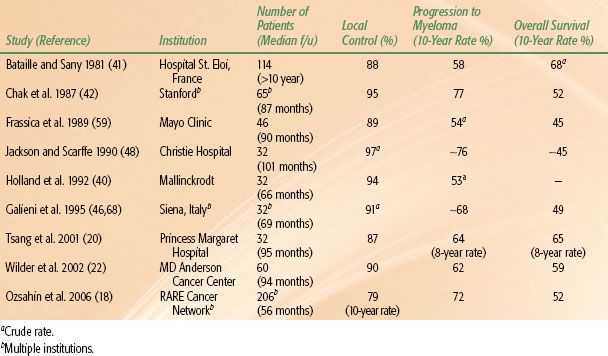
TABLE 81.5 SOLITARY EXTRAMEDULLARY PLASMACYTOMA: REPRESENTATIVE TREATMENT RESULTS (SERIES INCLUDING MORE THAN 15 PATIENTS)
Initial Treatment of Symptomatic Multiple Myeloma
Patients who have symptomatic multiple myeloma require treatment of the malignant plasma cell clone. Once the decision is made to treat, however, the first step is to determine candidacy for autologous stem cell transplantation (ASCT). As this modality has become the standard of care for eligible patients, it is necessary to stratify patients initially so that the ability to collect stem cells is not compromised by induction therapy.74
Patients Eligible for Autologous Stem
Cell Transplantation
In patients who are candidates for ASCT, various regimens can be used to induce response prior to stem cell collection. Historically, most regimens were steroid based, either with high-dose dexamethasone alone75 or with vincristine, Adriamycin (doxorubicin), and dexamethasone (VAD).76 Newer agents that have been validated in the relapse setting are now being used as initial therapy with superior results, including bortezomib and lenalidomide. Both have recently been established into initial treatment or for relapsed disease.
Bortezomib
Bortezomib was the first proteasome inhibitor to be used in clinical trials and has demonstrated efficacy and safety in frontline therapy when used in combination. Indeed, response rates have dramatically improved when compared with VAD or dexamethasone alone. A randomized comparison of bortezomib plus dexamethasone (BD) versus VAD as induction therapy before ASCT showed that BD produced superior complete response or near complete response: 14.8% versus 6.4%, at least very good partial response: 37.7% versus 15.1%, and overall response (78.5% vs. 62.8%) than VAD. Median progression-free survival was 36.0 months (BD) versus 29.7 months (VAD; P = .064).77 Even as a single agent, with dexamethasone or with doxorubicin, bortezomib has remarkable efficacy and safety in initial therapy.78,79 It is often the preferred agent in patients with renal insufficiency and high-risk disease. Its greatest challenge, however, remains neuropathy, occurring in 13% to 15% of patients at ≥grade 3; this may be reduced, however, with weekly use80 or when given subcutaneously.81
Lenalidomide
Lenalidomide is an immunomodulatory drug derived from thalidomide that has also been shown to be effective, both as upfront therapy and in relapsed disease. It is most commonly used in combination with low-dose dexamethasone.82 A phase III trial of lenalidomide with low-dose dexamethasone versus lenalidomide with high-dose dexamethasone found that despite a higher rate of complete or partial response with high-dose therapy, overall 1-year survival was 87% in the high-dose group versus 96% in the low-dose group (P = .0002), largely due to the significant toxicity of the former. As a result, the trial was stopped and patients on high-dose therapy were crossed over to low-dose therapy. Three-year overall survival rates now exceed 85%. This has resulted in the extensive use of this combination in upfront myeloma.82
Lenalidomide has also been used in combination with conventional chemotherapy and most recently with bortezomib.83 This has resulted in even higher response rates and complete remission rates of >50%.
Thalidomide
An alternative to VAD induction is the combination of thalidomide and dexamethasone (TD). Early reports indicate that this combination yields a response rate of 64% (similar to VAD), without compromising the ability to collect stem cells, but with a rate of deep vein thrombosis of 12%.84 The Medical Research Council Myeloma IX trial compared cyclophosphamide-thalidomide-dexamethasone (CTD) with cyclophosphamide-VAD as induction before ASCT. In a preliminary analysis, the complete response rate was 20.3% after CTD and 11.7% after cyclophosphamide-VAD.85
In a randomized trial of 480 patients, Cavo et al.86 reported that the addition of bortezomib to TD prior to tandem autologous stem cell transplant increased the complete or near-complete response rate to 31% versus 11% without bortezomib.
In summary, preferred initial regimens include bortezomib or lenalidomide, but alternatives include thalidomide or doxorubicin prior to ASCT.
Patients Not Eligible for Autologous Stem Cell Transplantation
In patients who will not be undergoing a transplant, there are various options available for initial therapy. Historically, most transplant-ineligible patients received melphalan and prednisone (MP), which produced partial remissions in approximately 55% of patients, with the occasional complete response.87 San Miguel et al.88 reported the results of a randomized trial of 682 patients randomized to receive either 9 6-week cycles of MP or the same chemotherapy with bortezomib. The addition of bortezomib increased the proportion of patients achieving and complete response (30% and 4%;, P <.001) and improved median duration of response (19.9 vs. 13.1 months), time to progression (24.0 vs.16.6 months; P <.001), and the risk of death (hazard ratio 0.61 for the bortezomib group; P = .008).
The addition of thalidomide to melphalan and prednisone (MPT) also improves outcome compared with MP alone. For patients aged 60 to 85, Palumbo et al.89 demonstrated a 76% response rate with MPT, superior to the 48% among patients treated with MP; however, thromboses were more common with thalidomide with an incidence of 12% (vs. 2% in the MP group).
In an updated analysis with median follow-up of 38.1 months, the median progression-free survival was 21.8 months for MPT and 14.5 months for MP (P = .004). The median overall survival was not significantly improved with MPT (45.0 vs. 47.6 months for MP, P = .79).90
In a randomized trial of 292 patients aged 75 years or older, Hulin et al.91 found that the addition of thalidomide to MP increased overall survival compared with MP alone (median survival 44.0 vs. 29.1 months; P = .028). Another randomized comparison of MPT versus MP among 357 elderly patients found that the addition of thalidomide increased the rate of good partial response or better (23% vs. 7%; P < .001) but did not improve progression-free or overall survival.92
A recent meta-analysis of MP versus MPT concluded that MPT increases response rates and overall survival, but with increased toxicity such as thrombosis and somnolence.93
These results provide several options for the initial therapy of patients who will not proceed to ASCT, including all three novel agents, thalidomide, bortezomib, and lenalidomide, with or without melphalan.
Autologous Stem Cell Transplantation
ASCT has become the standard of care for eligible patients, as it has been demonstrated in multiple trials to improve the likelihood of complete response, prolong disease-free survival, and extend overall survival.94–96 Treatment-related mortality rates are now <2%, and often the transplant can be performed entirely as an outpatient. Melphalan 200 mg/m2 is the most commonly used conditioning regimen, although it may be reduced in elderly patients or patients with renal insufficiency.
Tandem Transplantation
Tandem or double transplantation refers to a planned second ASCT after the patient has recovered from the first. A phase III trial in France evaluated tandem transplant versus single ASCT and demonstrated superior overall survival in the tandem group97; however, when further analyzed, the patients who benefited most from the second transplant were those who did not achieve a 90% reduction in their disease after the first ASCT. Therefore, it may be more prudent to consider tandem transplantation only in patients whose response to the first ASCT is suboptimal.
Allogeneic Stem Cell Transplantation
Myeloablative stem cell transplant is perhaps the only current potential cure for patients with myeloma, as the graft is not contaminated with tumor cells and may produce a profound graft versus myeloma effect.98 However, its use is very limited due to the lack of donors, age restriction, high treatment-related mortality, and graft versus host disease. Reduced intensity nonmyeloablative allogeneic transplant following ASCT has also been investigated as a means of inducing a graft versus myeloma effect. In one study of 102 patients undergoing nonmyeloablative transplant, 5-year overall survival and progression-free survival were 64% and 36%, respectively, although the 5-year rate of nonmyeloma mortality was 18%.99
Maintenance Therapy
Much investigation of late has been directed at the use of maintenance therapy post-ASCT to prolong remission and survival. Two large randomized trials are being conducted using maintenance lenalidomide versus placebo, with publication pending.100,101 In both trials progression-free survival was prolonged by approximately 20 months, although overall survival data are pending. The use of maintenance therapy post-ASCT remains controversial, and most guidelines do not recommend its use unless the patient is at high risk of rapid recurrence.
TABLE 81.6 TREATMENT OPTIONS FOR RELAPSED MULTIPLE MYELOMA





