Chapter 27 Pituitary Tumors
Etiology and Epidemiology
Etiology
The etiology of most pituitary tumors is not known and has been a source of debate. Based on two rat pituitary models, two primary modes of pituitary oncogenesis have been suggested.1 The first model, known as the hyperplasia-adenoma sequence, suggests a hormone-dependent pathway. Another model suggests de novo occurrence without the development of hyperplasia, possibly because of a genetic alteration. In this model, dysregulation of hormone or growth factor signaling leads to hyperplasia and tumor formation; the defect has been found especially in transgenic and knockout animal models, although there is strong evidence implicating overexpression of transforming growth factor–alpha (TGF-α), fibroblast growth factor (FGF), and fibroblast growth factor receptors (FGFRs) in tumor formation.2
For some patients with pituitary adenomas, a genetic predisposition has been described with four genes known to be associated with familial pituitary tumor syndromes: MEN1, CDKN1B, PRKAR1A, and AIP.3 Of patients with MEN1, which is an autosomal dominant disease characterized by tumors of the pancreatic islet cells, parathyroid glands, and pituitary gland, 40% will develop pituitary adenomas the great majority of which are prolactinomas. Pituitary tumors associated with the MEN1 syndrome have demonstrated loss of heterozygosity of chromosome 11q13 (locale of the MEN1 gene), which has been implicated in malignant progression of pituitary adenomas.4 Mutation of this gene does not appear to increase the risk for sporadic pituitary adenomas.5 In Carney’s complex, a rare inherited condition characterized by endocrine overactivity, schwannomas, abnormal skin pigmentation, and myxomas, the implicated genetic defect is loss of function of PRKAR1A, the gene for which is located on chromosome 17q23-24.6 Additional genetic alterations are found in McCune-Albright syndrome, a genetic disorder of bones, skin pigmentation, and hormones associated with premature puberty. Activating or gain-of-function GNAS1 mutations (of the guanine nucleotide-binding protein [G protein], alpha-stimulating activity polypeptide 1 gene on chromosome 20q13.1) are seen. Patients with McCune-Albright syndrome are present in the mosaic state, resulting from a postzygotic somatic mutation appearing early in the course of development that yields a monoclonal population of mutated cells within variously affected tissues. In isolated familial somatotropinomas (i.e., the occurrence of two or more cases of acromegaly in a family without a history of MEN or Carney’s complex), germline alterations in the aryl hydrocarbon receptor interacting gene (AIP gene) have been identified.7,8
Epidemiology
Although 10% of normal adults have a pituitary abnormality detectable on MRI, pituitary tumors account for only 10% to 15% of all diagnosed primary brain tumors. The frequency of pituitary adenomas varies greatly, although recent community-based studies from Belgium and the United Kingdom suggest that the prevalence is higher than has been historically noted.9,10
Based on a meta-analysis, the overall estimated prevalence of pituitary adenomas was 16.7%, with 14.4% observed in autopsy series and 22.5% in radiographic series.11 Over time, different criteria have been used for distinguishing hyperplasia from pituitary adenomas. The broader application of immunohistochemical staining for pituitary hormones and the differences in slice thicknesses for MRI may account for the wide variation in incidence that is reported in the literature.
Biologic Characteristics and Molecular Biology
Despite the screening of common oncogenes and tumor suppressor genes for pituitary adenomas, the initial transforming event for tumorigenesis has not been identified. Based on X chromosome inactivation analysis, it is apparent that most pituitary adenomas derive from a monoclonal expansion, which suggests that general principles of tumorigenesis may apply to pituitary adenoma formation.12 The MAPK (mainly Ras/extracellular signal-regulated protein kinase [ERK]) and the PI3K/Akt signaling pathways appear to have putative roles in pituitary tumorigenesis because activity of these pathways is increased with pituitary tumors. Although its role is not fully understood, a docking protein in the endothelial growth factor receptor (EGFR) pathway, EGF pathway substrate number 8 (Eps8), is overexpressed (5.9 times higher) in pituitary tumors compared with normal anterior pituitary gland tissue.13 Because pituitary tumors are mostly benign, a protective mechanism to restrict uninhibited growth of tumor cells is probably active. The concept of oncogene-induced senescence is one possible mechanism. High pituitary p21Cip1/WAF1 levels appear to promote senescence and restrict tumor growth.14
For somatotroph (growth hormone) adenomas, an activating mutation of the alpha subunit of the guanine nucleotide stimulatory protein (Gs-alpha) gene is found in about 40% of patients, which results in activation of adenylyl cyclase.15 Overexpression of the pituitary tumor transforming gene (PTTG) is also seen in most human somatotroph adenomas.16 Mutations of fibroblast growth factor–4 (FGF-4) have led to development of lactotroph adenomas in transgenic mice.17
Anatomy, Pathways of Spread, and Pathology
Anatomy and Pathways of Spread
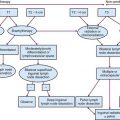
The pituitary gland is a midline intracranial organ that measures approximately 8 mm in the anteroposterior axis, 10 mm in the transverse axis, and 6 to 8 mm in the superior-inferior axis. It occupies the cavity in the sphenoid bone known as the sella turcica. The pituitary gland is separated from the structures above it (i.e., the optic chiasm, hypothalamus, anterior cerebral arteries, and floor of the third ventricle) by the sellar diaphragm, which is formed by a circular fold of dura. Immediately anterosuperior to the diaphragm sella lies the optic chiasm. The pituitary stalk (infundibulum) crosses the sellar diaphragm and connects the hypothalamus to the pituitary gland. Anteriorly, the tuberculum sellae lies in the floor of the sella and extends laterally to form the anterior clinoid processes. The posterior aspect of the sella forms the dorsum sellae and extends laterally to form the posterior clinoid processes. Laterally, the cavernous sinuses contain cranial nerves III, IV, V1 (ophthalmic nerve), V2 (maxillary nerve), and VI and the internal carotid artery. Given the proximity of the pituitary to these various structures, parasellar tumors can affect cranial nerves III, IV, V, and VI, and suprasellar tumor extension can lead to bitemporal hemianopsia via compression of the optic chiasm. Figure 27-1 demonstrates the complex anatomy of the parasellar region.
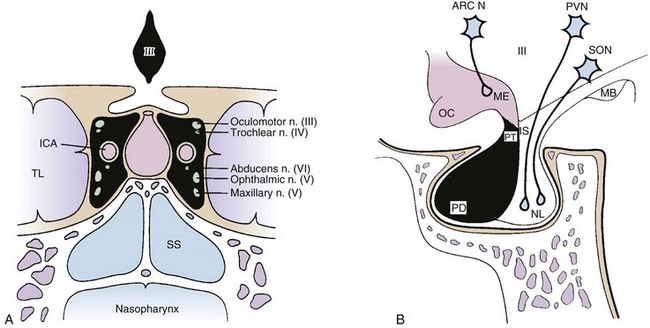
The pituitary has two distinct embryologic origins; the first gives rise to the anterior and intermediate pituitary lobes, and the second gives rise to the posterior pituitary lobe. The anterior lobe (adenohypophysis) and intermediate lobe are derived from Rathke’s pouch, which is an evagination of ectodermal tissue from the primitive oral cavity (pharyngeal pouches). The posterior lobe (neurohypophysis) and stalk are derived from the diencephalon and, therefore, have a neuroectodermal origin. The anterior pituitary lobe accounts for most of the pituitary gland and produces at least six hormones: prolactin (PRL), adrenocorticotropic hormone (ACTH), follicle-stimulating hormone (FSH), luteinizing hormone (LH), growth hormone (GH), and thyroid-stimulating hormone (TSH).18 The intermediate lobe produces melanocyte-stimulating hormone (MSH). The posterior lobe stores and releases two hormones, vasopressin (antidiuretic hormone [ADH]) and oxytocin, which are produced by the hypothalamus.
Pituitary tumors are generally classified as microadenomas or macroadenomas depending on the diameter of the tumor as described by Hardy.19 A microadenoma is a tumor less than 1 cm in diameter, whereas a macroadenoma has a diameter of 1 cm or greater. Incidental microadenomas have been observed with a frequency of 3% to 25% in large, unselected autopsy series.20 Microadenomas are more frequently seen in females, whereas macroadenomas occur with equal frequency in males and females. Macroadenomas are more common than microadenomas. For lesions less than 3 mm in diameter, the term picoadenoma has been used.21
Pathology
According to the World Health Organization (WHO) classification of pituitary tumors, these tumors are defined as neoplasms located in the sella turcica.22 This classification is based on structural similarities of the normal parenchymal cells and the immunohistochemical demonstration of hormone secretion. As a result, pituitary adenomas may express more than one or two hormones. The classification includes a new entity designating a borderline adenoma or adenoma of uncertain behavior.22 The atypical adenoma is defined as an invasive tumor with an elevated mitotic index (MIB-1-labeling index greater than 3%) and extensive nuclear immunostaining for TP53.22
Most lesions are easily identified on routine histopathologic testing, but metastatic well-differentiated neuroendocrine carcinoma can present a diagnostic challenge.23 For patients with acromegaly or Cushing’s disease, hyperplasia may be clinically indistinguishable from an adenoma. Differentiation of hyperplasia from adenoma can be made using a reticulin stain. Normal pituitary tissue is composed of small acini of pituitary cells surrounded by an intact reticulin network. In hyperplasia, the architecture of the acini is maintained, but the acini are increased in size, and the reticulin stain demonstrates an intact network. Pituitary adenomas are characterized by a complete disruption of the reticulin fiber network.
The pathologic hallmark of pituitary adenomas is the monotonous and monomorphous proliferation of neoplastic cells that replace the normal acinar pattern in the pituitary lobe. Growth hormone–producing cells are seen with greater frequency anteriorly in the lateral aspects of the pituitary. Prolactin-producing cells are distributed throughout the pituitary but have a greater density in the posterior lateral aspect of the gland. Adrenocorticotropic hormone–producing cells are present in the median wedge. Thyroid-stimulating hormone–producing cells are seen in the anterior aspect of the median wedge. Gonadotropin-producing cells are distributed throughout the anterior pituitary. Nonfunctioning adenomas typically have solid sheets, nests, and sinusoidal patterns and are interrupted by pseudopapillae and striking pseudorosettes around vascular channels. Overlap of the distribution of these functional hormonal secretory cells does exist, and they do not reside in well-demarcated zones of the pituitary. Mitoses are seen in 3.9% of noninvasive adenomas, 21.4% of invasive adenomas, and 66.7% of carcinomas.24
The usefulness of labeling-index and TP53 immunostaining is not clear, because these assessments may not correlate with tumor behavior. Invasive pituitary adenomas usually exhibit a higher Ki-67 proliferation index.25 Markers such as proliferating cell nuclear antigen (PCNA), Ki-67/MIB-1, and antiapoptotic Bcl-2 have not demonstrated consistent correlation with tumor invasiveness or recurrence.26 Galectin-3, a β-galactoside-binding protein, may play a role in pituitary tumor progression, and immunostaining for this is also available.27
The pituitary is composed of at least six distinct cell types, which are responsible for the production of at least one hormone. Advances in molecular biology have identified three major pathways (T pit; Pit-1; and SF-1, GATA-2, ER) of cytodifferentiation of adenohypophyseal cells.28,29 These pathways are determined by a complex pattern of transcription expression, which can help classify the adenomas.30 In situations where histologic and immunohistochemical profiles are atypical, electron microscopy can be helpful in achieving accurate classification.
Clinical Manifestations, Patient Evaluation, and Staging
Clinical Manifestations
Patients with pituitary tumors can have a number of different symptoms and presentations, depending on the size and mass effect of the tumor and hormonal abnormalities. Headaches can occur with a lesion of any size. When the tumor extends superiorly into the suprasellar region, visual loss can occur, including bitemporal or homonymous hemianopsia, superior or inferior field cut deficits, and central scotoma. Extension into the cavernous sinus can lead to cranial nerve dysfunction (commonly, palsies of cranial nerves III and IV). Cranial nerves V and VI may also be affected. Lateral extension into the temporal lobe may cause seizures. The clinical presentations and endocrine abnormalities can vary, depending on the age and sex of the patient. Patients with large adenomas may have compromised pituitary function, which can lead to hypogonadism, secondary hypothyroidism, and adrenal insufficiency.31 Diabetes insipidus is extremely rare and most commonly is a manifestation of an invasive and infiltrative tumor (such as a metastasis), an inflammatory condition (such as neurosarcoidosis), or a lesion that arises from or is attached to the infundibulum or hypothalamus (such as a craniopharyngioma).32 If the gland acutely infarcts or hemorrhages, pituitary apoplexy may result, which may require urgent surgical decompression.
Patient Evaluation
Laboratory Assessment
Initial tests can determine a pituitary deficiency and diagnose the secretory status of the adenoma. Screening studies should include thyroid-stimulating hormone, free thyroxine (T4), adrenocorticotropic hormone, cortisol, prolactin, somatomedin C (insulin-like growth factor–1 [IGF-1]), luteinizing hormone, follicle-stimulating hormone, alpha subunit, and, in men, testosterone. In addition to endocrine evaluation, a complete blood count, blood chemistry assessment, and urinalysis should be obtained. Because physiologic hormonal variations in the blood and urine levels can occur, the interpretation of these results should be considered based on diurnal variations, age and gender of the patient, and pregnancy and menopausal status. The conditions and timing under which the samples are obtained also influence interpretations of the results. Because the diagnosis of Cushing’s disease can be difficult, multiple tests performed over time and at different times during the day may be needed for this diagnosis to be conclusive.33
For patients with prolactinoma who also have macroadenoma, the prolactin level is usually greater than 200 ng/mL. Other common conditions that have been associated with hyperprolactinemia include end-stage renal disease, renal insufficiency, depression, primary hypothyroidism, acquired immunodeficiency syndrome, sarcoidosis, and nonalcoholic cirrhosis.34 On the other hand, pituitary stalk compression from tumor may also cause elevated prolactin levels in the range of 150 ng/mL or less. Because antidepressants, protease inhibitors, verapamil, and phenothiazines may elevate prolactin levels, carefully review the medications that the patient is taking. A pregnancy test is mandatory for women with amenorrhea or hyperprolactinemia. Elevated serum prolactin levels greater than 300 ng/mL are usually diagnostic of pituitary adenoma, and levels higher than 100 ng/mL in nonpregnant patients often are associated with pituitary adenoma.
For patients with acromegaly, most cases result from excess secretion of growth hormone by a pituitary tumor.35 The definitive test is measurement of growth hormone response to 75 or 100 g of oral glucose (oral glucose tolerance test). This glucose load should normally cause marked suppression of the growth hormone release to a level below 2 ng/mL. To ensure accuracy of the test, these measurements of serum glucose and growth hormone must be taken every 30 minutes for 2 hours. The IGF-1 value is also elevated after adjusting for sex and age variation. IGF-1 levels provide the best intermittent method for monitoring response to treatment, although dynamic testing of growth hormone kinetics with the oral glucose tolerance test (OGTT) is also frequently used.
Imaging
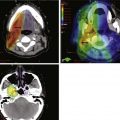
MRI of the brain with gadolinium is the imaging test of choice, given its superior resolution compared with CT scanning (Fig. 27-2). Thin slices (at 2- to 3-mm intervals) obtained before and after gadolinium contrast administration with images in the coronal, axial, and sagittal planes provide detailed information for the initial diagnosis and allow detection of small lesions. The posterior lobe of the pituitary has a high signal intensity on T1-weighted images (posterior pituitary bright spot) that distinguishes it from the anterior lobe, which has a signal intensity similar to that of white matter. Dynamic coronal imaging techniques after contrast administration enhance normal pituitary tissue earlier and more intensely and help delineate adenoma tissue, which tends to enhance later. For patients who have undergone previous surgery, fat suppression techniques can help differentiate surgical fat grafts from tumor tissue. For patients undergoing stereotactic radiosurgery (SRS), thin-slice (1-mm) imaging with contrast medium is obtained to define the tumor and optic apparatus. For hypersecretory adenomas, positron emission tomography (PET) imaging with co-registration may be valuable.36
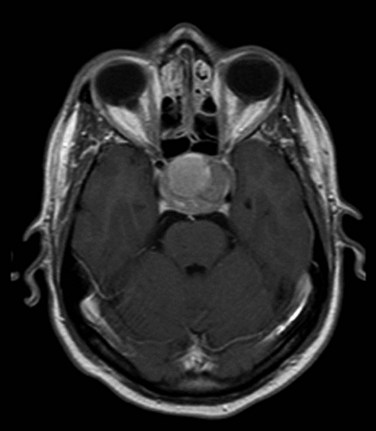
For patients with suspected Cushing’s disease, thin-slice images of 1-mm thickness have greater sensitivity; even so, the tumor may not be detected in about 50% of patients.37 Spoiled gradient recalled acquisition sequences may have superior sensitivity (80%) compared with conventional spin echo images following contrast enhancement.38 The degree of contrast enhancement does not differentiate one sellar mass from another. A clear distinction between an intrasellar mass and normal pituitary tissue is not consistent with a pituitary adenoma.
Staging
Classification of detectable or symptomatic pituitary tumors can be based on endocrine function, clinical presentation, and anatomic extent of disease. These tumors can also be broadly classified as functional or nonfunctional based on their secretory activity. As adenomas enlarge, they can extend into suprasellar, parasellar, or infrasellar structures. To aid with surgical and imaging assessment, Hardy and Verzina39 developed a classification system. Wilson’s40 modification of this classification system incorporates imaging and intraoperative findings of sellar destruction (grade) and extrasellar extension (stage). Others have defined giant adenoma as a lesion with extension beyond the sella and suprasellar space or size greater than 4 cm.41,42 No universally accepted staging system exists for pituitary tumors.
Primary Therapy
Surgery
Advances in transsphenoidal approaches with microsurgical techniques have made this procedure the initial treatment of choice for patients with nonfunctioning pituitary adenomas, acromegaly, and Cushing’s disease. Surgery provides immediate decompression for patients with progressive visual loss or pituitary hemorrhage. For patients with prolactinoma who do not tolerate or respond to medical therapy, surgery can be considered. Surgery has also been useful in reducing the tumor bulk for combined management with radiation therapy or medical therapy. For patients undergoing SRS, surgery may play an important role because a distance of 2 to 5 mm is needed between the tumor and the optic apparatus to deliver a sufficient dose to the tumor and to minimize the risk for optic neuropathy. Reoperation results in lower rates of success compared with the initial surgery.43
The transsphenoidal approach has become less invasive with development of a newer approach, endoscopic, minimally invasive pituitary surgery (MIPS). This technique can result in shorter hospitalization times.44,45 With MIPS, nasal or intraoral incisions are not needed and nasal speculums and nasal packing need not be used. There appears to be a decrease in complication rates compared with traditional sublabial transseptal approaches. MIPS is performed jointly with otorhinolaryngologists. It may also be used to complement microsurgical approaches.
Based on a questionnaire study of 958 neurosurgeons reporting on their own experience with transsphenoidal surgery, the risk for complications was inversely proportional to the surgeon’s level of experience.46 Reported complication rates among experienced surgeons include perioperative mortality (<1%), central diabetes insipidus (2%), cerebrospinal rhinorrhea (2%), and meningitis (2%). Transient central diabetes insipidus is very common and reported to occur in 22% of patients at a very active pituitary surgery center.47 These complications are more common in patients with macroadenomas. Less experienced surgeons are more likely to cause the above complications.45,48
Medical Therapy
Medical therapy can be used in several situations for patients with growth hormone–secreting tumors. Somatostatin analogs such as octreotide or lanreotide can be used to lower elevated growth hormone levels before surgery, for patients with persistent growth hormone and IGF-1 elevation after surgery, and for patients who continue to receive medical therapy until radiation therapy or radiosurgery has taken full effect. Pegvisomant, a growth hormone receptor antagonist, appears to be the most effective medical therapy for IGF-1 normalization.49 The cost of this therapy is, however, substantial (>$50,000 per year).
Medical treatment of hormonal deficiencies resulting from the pituitary tumor is best managed by an endocrinologist. Hypopituitarism can have variable clinical manifestations, and management should be individualized to minimize the impact of this condition. Depending on the deficiency, glucocorticoids, gonadal steroids, and thyroid hormones may need to be initiated. Commonly used medicines include hydrocortisone and cortisone acetate for glucocorticoid replacement and L-thyroxine for hypothyroidism. Gonadal steroids used include estrogen and progestin for women and testosterone for men. Careful optimization of medication supplementation, especially to avoid supraphysiologic doses of steroids, appears to be important in minimizing the effect of hypopituitarism.50
Radiation Therapy and Stereotactic Radiosurgery
The lack of prospective, randomized data comparing different radiation delivery methods has resulted in strong proponents for fractionated versus radiosurgical approaches.51,52,53 Most of the literature is based on single institutional results .
.
Advances in radiation therapy have led to modern radiation techniques that allow more focused delivery of radiation to the tumor and decreased dose to the normal brain. Sophisticated treatment planning systems, incorporation of CT and MRI studies, stereotactic guidance, radiation approaches such as intensity-modulated radiation therapy and image-guided radiation therapy, micro-multileaf collimators, and relocatable frames or masks have improved precision.53,54,55 Results with radiation therapy, including specifics regarding dosing and margins, will be discussed in greater detail later in the chapter.
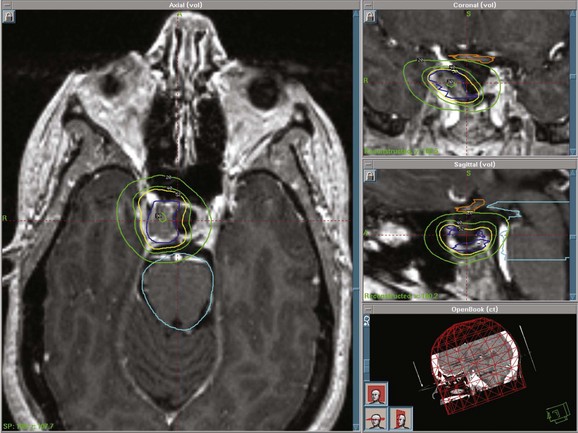
Stereotactic radiosurgery (SRS) delivers a highly focused, single, large fraction of ionizing radiation to a small intracranial target volume (Fig. 27-3). Frame-based systems (e.g., proton beam, helium ion, Gamma Knife, and linear accelerator SRS techniques) and frameless systems (e.g., CyberKnife and linear accelerator-based units) have been used. Given the sharper dose gradient of SRS versus fractionated approaches, the potential for radiation injury to normal brain and vessels and secondary tumor formation may be less with SRS.


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


