div class=”ChapterContextInformation”>
3. The Pituitary Gland
3.1 Anatomy and Embryology
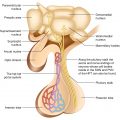
The pituitary gland comprises the predominant anterior lobe (adenohypophysis), the posterior lobe (neurohypophysis), and a vestigial intermediate lobe. The anatomic relationships between the pituitary and the main nuclei of the hypothalamus were already described earlier.
The anterior pituitary (adenohypophysis) originates from Rathke pouch, an ectodermal evagination of the oropharynx, and migrates to join the neurohypophysis. The portion of the Rathke pouch in contact with the neurohypophysis develops less extensively and forms the intermediate lobe. The posterior lobe of the pituitary (neurohypophysis) is of neural origin, arising embryologically as an evagination of the ventral hypothalamus and the third ventricle, and is made up of the endings of neurons whose cell bodies reside in the supraoptic and paraventricular nuclei of the hypothalamus, as already specified earlier.
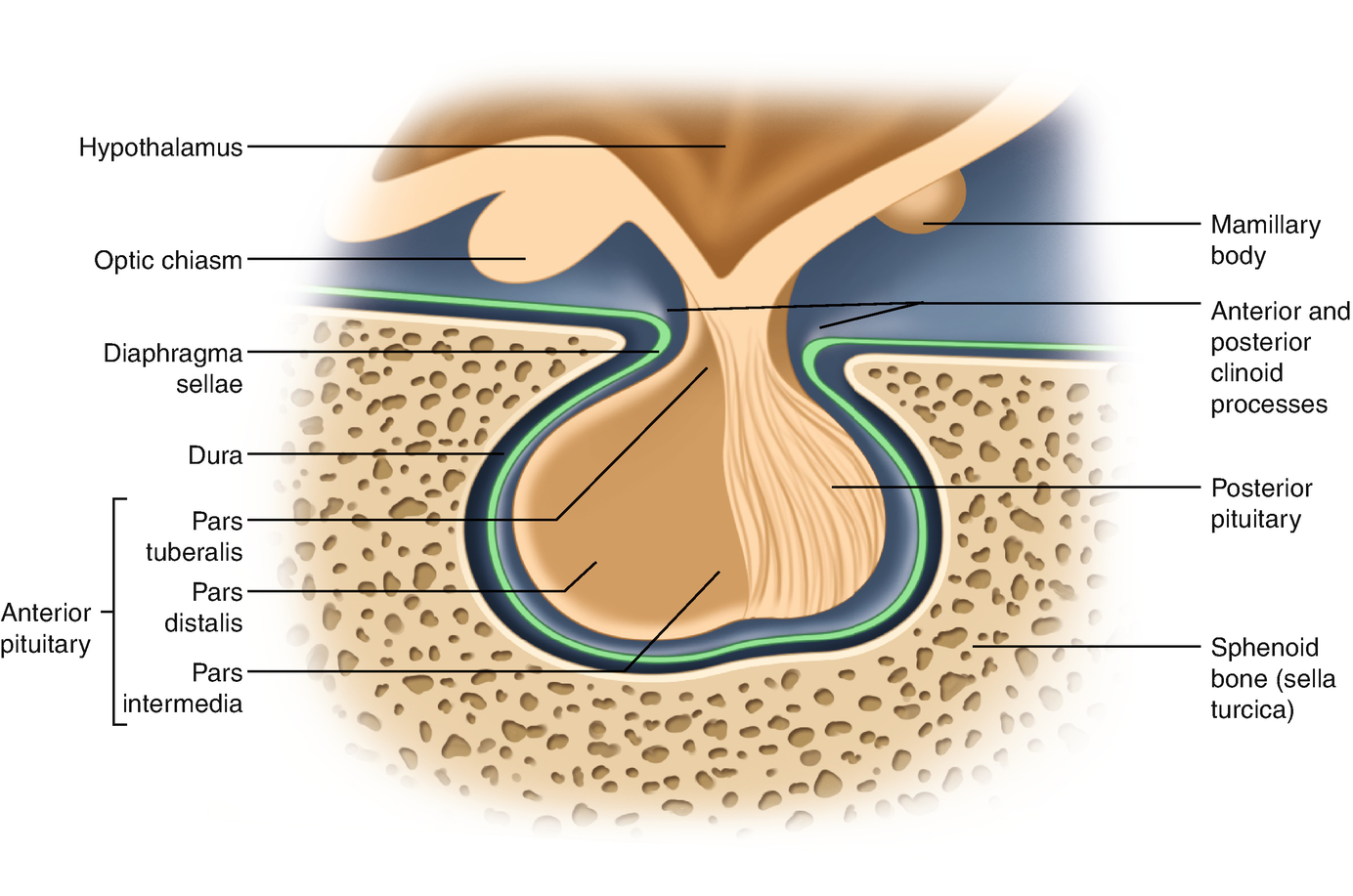
Representation of the normal relationships of the pituitary gland
The pituitary measures around 13 mm transversely, 9 mm anteroposteriorly, and 6–9 mm vertically and weighs approximately 500–900 mg.
3.2 Adenohypophysis (Anterior Pituitary)
3.2.1 Major Adenohypophyseal Hormones and Their Cellular Sources
Major adenohypophyseal hormones and their main functions
GH | • Stimulates the production of IGF-1 (the mediator of the indirect actions of GH) • Also exerts direct actions on growth and metabolism |
PRL | • Stimulates milk production (protein and lactose synthesis, water excretion, and sodium retention) • Inhibits gonadotropins secretion |
ACTH | • Stimulates glucocorticoids and sex steroids production in the zona fasciculata and zona reticularis of the adrenal cortex, inducing hyperplasia and hypertrophy of the adrenal cortex |
TSH | • Stimulates all aspects of thyroid gland function: hormone synthesis, secretion, hyperplasia, hypertrophy, and vascularization of the thyroid gland |
LH | • Females: stimulates steroid hormones (androgens) synthesis in theca interna cells, lutein, and hilar cells; ovulation induction; promotes luteinization, maintains the corpus luteum, stimulates the corpus luteum to produce progesterone • Males: stimulates steroid hormones production in Leydig cells |
FSH | • Females: targets the granulosa cells to promote follicular development; stimulates aromatase expression and inhibin secretion • Males: targets the Sertoli cells to promote spermatogenesis and to stimulate inhibin secretion |
3.2.2 The Pituitary Tumoral Syndrome (The Pituitary Mass Effects)
3.2.2.1 Definition
The pituitary tumoral syndrome includes the sum of symptoms and signs caused by an expanding pituitary mass into the sella turcica or into the suprasellar space (the space between hypothalamus-hypophysis).
3.2.2.2 Etiology
Pituitary Adenomas
Over 95% of cases are benign and most are monoclonal.
In adults, the most common cause of hypothalamic–pituitary dysfunction is a pituitary adenoma, of which the great majority is hypersecreting.
Pathogenesis
The mechanism of pituitary tumor genesis remains largely unclear. Pituitary adenomas are monoclonal, supporting the theory that there are intrinsic molecular events leading to pituitary tumor genesis.
- 1.
According to the size of the tumor
- (a)
Microadenomas: Intrasellar adenomas <1 cm in diameter, without sellar enlargement or extrasellar extension
- (b)
Macroadenomas: >1 cm and causing generalized sellar enlargement
- (a)
- 2.
According to the state of hormone secretion
- (a)
Nonfunctional pituitary adenoma
- (b)
Functional pituitary adenomas: for example, prolactinomas
- (a)
Craniopharyngioma
Features
Raised intracranial pressure
Visual disturbance
Hypothalamic–pituitary disturbance
Growth failure in children
Precocious puberty and tall stature are less common
Anterior and posterior pituitary failure, including diabetes insipidus
Weight gain
Metastatic Lesions
Carcinomas that metastasize to the hypophysis include breast, lung, and gastrointestinal cancers.
Miscellaneous Lesions
Such as sarcoidosis, histiocytosis x, lymphomas, lymphocytic hypophysitis, pituitary abscesses, meningioma, and cysts.
3.2.2.3 Clinical Features
- 1.
Neurological signs
- (a)
Headache = the major symptom, with some specific characteristics: frontal, frontoparietal or retro-orbital localization; none or only partial amelioration with the usual painkillers drugs administration; progressive in severity.
Note: The substratum of a headache is represented by diaphragmatic distortions or dural impingement (stretching of the dural plate) by a tumoral mass having an anterior—superior evolution.
- (b)
The evolution of pituitary masses toward the hypothalamus can cause many other abnormalities, including disorders of consciousness, behavior, thirst, appetite, and temperature regulation; the typical features are hyperphagia and weight gain, loss of thirst sensation, diabetes insipidus, somnolence, problems with temperature control, and behavior change (the hypothalamic syndrome); these abnormalities are usually accompanied by hypopituitarism.
- (c)
Large pituitary lesions may extend laterally into the cavernous sinus, compromising the function of the third, fourth, or sixth cranial nerve, leading to diplopia, palpebral ptosis, ophthalmoplegia and internal carotid artery compression, or cavernous sinus thrombosis.
- (d)
Very large tumors with both suprasellar and parasellar extension may lead to symptoms of increased intracranial pressure (↑ ICP) such as severe, persistent headache, vomiting, seizures, bradycardia, focal neurology, papilledema = neurosurgery emergencies.
- (a)
- 2.
Ophthalmological signs = neurosurgery emergencies
Impinging upon the optic chiasm or optic tract results in the optic—chiasmatic syndrome.
Because of the anatomy of the chiasma, pressure from below affects temporal visual fields, starting superiorly (pressure on the inferior crossing chiasmatic fibers which lead to bitemporal visual loss, in the superior field portions = bitemporal sup quadrantanopia) and ultimately extending to entire temporal field loss (bitemporal hemianopia).
Tumor evolution toward an optic tract leads to visual loss in the temporal field of one ocular globe and, respectively, the nasal field of the other ocular globe (homonymous contralateral hemianopia).
Rarely, decreased visual acuity, pupillary abnormalities, optic atrophy, papilledema, cranial nerve palsies, and nystagmus may be encountered.
- 3.
Endocrine and metabolic signs include symptoms and signs of hormone hypersecretion and/or hypopituitarism caused by tumoral local pressure on the healthy pituitary gland.
Pituitary Hypersecretion
Prolactinoma—the most common type, accounting for about 57% of primary pituitary tumors leads to amenorrhea, galactorrhea, infertility in ♀ and ↓ libido, impotence, and male infertility in ♂.
GH/GH+PRL secreting pituitary adenomas (10–15%)—leads to the more characteristic syndromes of acromegaly and gigantism.
ACTH-secreting pituitary adenomas (5%)—leads to the more characteristic syndromes of Cushing disease.
TSH-secreting pituitary adenomas (1%)—hyperthyroidism with goiter.
(FSH, LH) gonadotropin-secreting pituitary adenoma (1–2%)—usual large chromophobe adenomas presenting with visual impairment, hypopituitarism/rarely ovarian hyperstimulation.
Nonfunctioning pituitary adenomas—28%.
Pituitary Insufficiency
Pituitary failure (hypopituitarism) refers to either partial or complete deficiency of pituitary hormones (Table 3.2).
Main manifestations of hypopituitarism
Hormone deficiency | Clinical features |
|---|---|
GH | Children: short stature/adults: reduced exercise capacity, ↓ lean body mass, ↑ body fat, impaired psychological well-being,↑ cardiovascular risk |
LH/FSH | • ♀: Oligo/amenorrhea, anovulatory cycles, loss of axillary and pubic hair, osteoporosis • ♂: erectile dysfunction, ↓ libido, testicular atrophy, loss of sexual hair, osteoporosis |
TSH | As in primary hypothyroidism, except lack of goiter (see Sect. 3.6) |
ACTH | As in Addison’s disease (primary ACI) except lack of hyperpigmentation, hyperkalemia |
Prolactin | Failure of lactation |
ADH | Polyuria and polydipsia |
3.2.2.4 Imaging Studies
Magnetic resonance imaging (MRI) currently provides the optimal imaging of the pituitary gland and hypothalamus.
Computed tomography (CT) scans may still be useful in demonstrating calcification in tumors (e.g., craniopharyngiomas) and hyperostosis in association with meningiomas or evidence of bone destruction as well as for the discovery of hemorrhagic lesions.
Plain skull radiography (or Radiography of the sella turcica) may show evidence of pituitary fossa enlargement (however, it does not “see” the pituitary gland or a pituitary tumor) but has been superseded by MRI.
Magnetic Resonance Imaging (MRI)
Microadenomas: (2–9 mm)—On T1-weighted images, they appear as low-signal intensity lesions than the remainder of the normal gland and do not usually enhance with gadolinium (hypodense); sometimes difficult to be visualized. Contrast enhancement, asymmetry of the gland (a unilateral convex superior gland margin), or stalk position (e.g., deviation of the pituitary stalk toward the side opposite the adenoma) can be helpful in demonstrating their presence.
Macroadenomas: >10 mm in diameter—readily visualized with MRI scans, and the scan will also define the adjacent structures and the degree of extension of the lesion → compression of the normal pituitary and distortion of the pituitary stalk, compression, and upward displacement of the optic chiasma (suprasellar extension), cavernous sinus invasion (lateral extension), or sphenoid sinus extension.
Note: Contrary to microadenomas, macroadenomas, which are significantly more vascular, have a higher affinity to gadolinium.
MRI distinguishes pituitary adenomas from other masses.
Craniopharyngiomas—appear as intra- and/or suprasellar masses with cystic and/or solid components. Calcification is present in 45–57%, better visualized by CT or plain skull X-ray.
3.2.2.5 Management of Pituitary Masses
Excision of mass lesions causing pressure effects
Suppression/correction of hormonal hypersecretion
Preserving the normal secretion of anterior pituitary hormones (or correcting their insufficiency)
Pituitary adenomas are treated with surgery, irradiation, or drugs to suppress hypersecretion by the adenoma or its growth.
Surgical Treatment
Transsphenoidal microsurgical approach to the sella turcica, (endoscopically) = is the procedure of choice (with selective resection of the lesion).
In the transsphenoidal procedure, the surgeon approaches the pituitary from the nasal cavity through the sphenoid sinus, removes the anterior–inferior sellar floor, and incises the dura. The adenoma is selectively removed; normal pituitary tissue is identified and preserved.
Pituitary microadenoma: complete removal
Pituitary macroadenoma: partial excision
↓ morbidity and mortality
Major complications include postoperative hemorrhage, cerebrospinal fluid leak, meningitis, and visual impairment, transitory diabetes insipidus: <5% of patients.
Transfrontal/transparietal craniotomy
Transfrontal/transparietal craniotomy is required only for the rare invasive suprasellar mass extending into the frontal or middle cranial fossa or optic nerves or having extensive posterior clivus invasion.
! Accompanied more frequently by major neurological, endocrine complications (diabetes insipidus, pituitary failure), visual impairments.
Radiotherapy
Possibilities:
- 1.
Conventional radiation therapy (RT) using high-energy sources (total dose = 10,000–12,000 Rads in two to three sessions) is most commonly employed.
- 2.
Gamma knife radiosurgery utilizes stereotactic CT-guided cobalt 60-gamma radiation to deliver high radiation doses to a narrowly focused area while minimizing the mass of normal brain exposed to radiation. (An adequate distance of the pituitary mass from the optic chiasm is needed to prevent radiation-induced damage). Remission rates have been reported in the range of 43–78%.
Side effects: visual damage (damage to the optic nerve or chiasma), hypopituitarism (is common, and the incidence increases with time following radiotherapy—about 50–60% at 5–10 years), seizures, cognitive disorders, radionecrosis of brain tissue.
Medical Treatment
Suppression of hormone hypersecretion became feasible with the availability of bromocriptine or cabergoline, two dopamine agonists that suppress both PRL and tumor growth in patients with prolactinomas (sometimes used also in nonfunctioning pituitary adenomas), of somatostatin analogs, useful in the therapy of acromegaly, some TSH-secreting adenomas, Cushing’s disease (see later).
Replacement treatment of the anterior pituitary gland insufficiency.
3.3 Neurohypophysis (Posterior Pituitary)
Oxytocin
Prolactin and oxytocin are the hypothalamic/pituitary hormones critical to lactation.
In ♀, oxytocin stimulates contraction of the myoepithelial cells of the mammary ducts and the ejection of milk as well as uterine contraction during labor.
Milk secretion requires the additional stimulus of suckling, which activates a neural arc.
Suckling at the breast stimulates mechanoreceptors or tactile receptors that ascend through the spinal cord to the lateral cervical nucleus and eventually to the oxytocinergic magnocellular neurons in the SON and PVN. Neurotransmitters trigger the release of oxytocin, which is released in a pulsatile fashion, stimulating the cells along the duct to shorten and the ducts to widen ⇒ thus producing a pumping action on the alveoli and promoting maximal emptying of milk from the alveoli. Oxytocin release is occasioned by stimuli of a visual, psychologic, or physical nature that prepare the mother for breastfeeding, whereas PRL release is limited to the suckling reflex arc.
Oxytocin is the strongest stimulant to myometrial contraction, explaining its value as a therapeutic agent in inducing parturition and the interest in oxytocin antagonists to delay parturition. In humans, there is a dramatic ↑ in uterine response to oxytocin as parturition approaches. The most specific role of oxytocin may be the release of oxytocin brought about by vaginal and cervical dilatation produced by descending head and body. This may be important in stimulating the uterine muscle to contract maximally and clamp down blood vessels to decrease blood loss. Oxytocin infusion is commonly used to induce or augment labor. Both maternal and fetal oxytocin levels ↑ spontaneously during labor, but neither has been convincingly shown to trigger labor and, oxytocin deficiency has no known adverse effect on parturition or breastfeeding. At term, ↑ sensitivity of uterine tissue to oxytocin is attributed to ↑ synthesis and density of oxytocin receptors. Clinical studies using the oxytocin receptors inhibitor, atosiban, have demonstrated that delivery can be delayed by 24–48 h, but meta-analyses do not show much clinical improvement in neonatal outcome.
Oxytocin has no known role in ♂. It may aid contraction of the seminal vesicles.
ADH (AVP)
Vasopressin is the water-retaining hormone in all mammals and along with thirst is the primary regulator of osmolality. The release of vasopressin occurs in response to changes in osmolality detected by osmoreceptors in the hypothalamus and also large changes in blood volume ↓. Exercise, nicotine, angiotensin II, and high temperature also rise vasopressin secretion. On the contrary, low temperature (coldness) and alcohol decrease ADH secretion.
The main site of action of vasopressin is in the collecting duct and the thick ascending loop of Henle where it increases water permeability so that solute-free water may pass along an osmotic gradient to the interstitial medulla. Vasopressin in higher concentrations has a pressor effect. Therefore, ADH is an important regulator of plasma osmolality, water balance, and arterial pressure.
3.3.1 Diabetes Insipidus
Clinical features are the result of failure to secrete adequate quantities of ADH or the failure of the kidney to respond to circulating ADH. Diabetes insipidus (DI) is literally the excretion of a large volume of urine (diabetes) that is hypotonic (osmolality <300 mOsm/kg), dilute, and tasteless (insipid).
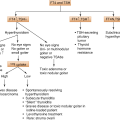
3.3.1.1 Classification of DI
According to the site of lesion:
Central DI
Nephrogenic DI
According to duration:
Transitory DI
Permanent DI
3.3.1.2 Etiology of DI
Central diabetes insipidus
Craniopharyngiomas (the most common solid tumor to produce DI), pituitary infiltration by metastases, suprasellar pinealomas, and germinomas in children
Surgery in the hypothalamic, pituitary stalk area
Head trauma
Infiltrative disorders: sarcoidosis, histiocytosis, lymphomas
Infections: meningitis, meningoencephalitis
Autoimmune
DIDMOAD syndrome = DI + diabetes mellitus (DM) + optic atrophy + deafness
Idiopathic
Nephrogenic diabetes insipidus
Acquired: chronic renal disease: any renal disease that interferes with collecting duct or medullary function (e.g., chronic pyelonephritis, toxic nephritis, postobstructive uropathy).
Hypokalemia, hypercalcemia.
Drugs: lithium, demeclocycline.
Familial: X-linked recessive—vasopressin receptor gene or autosomal recessive—aquaporin-2 gene
3.3.1.3 Clinical Features of DI
Adults—polyuria, nocturia, and thirst, polydipsia
Children—polyuria, enuresis and failure to thrive
Rarely: acute dehydration characterized by arterial hypotension, shock, and coma.
3.3.1.4 Investigations of DI
Diagnosis of DI Type
- 1.
Confirm large urine output (>3 l/day) (with low urinary density (the urine specific gravity) <1005)
- 2.
Exclude diabetes mellitus and renal failure (osmotic diuresis) as well as other causes of polyuria (hypokalemia and hypercalcemia: check electrolytes)
- 3.
Dehydration test (fluid deprivation test):
- (a)
No fluids for 12–18 h; measure body W, TA, pulse, urine flow, and urine-specific gravity each hour; urine and plasma osmolality
Note: In DI the urine flow remains increased, while urine specific gravity is less than 1005 and urine osmolality remains <300 mOsm/kg; the patient becomes dehydrated.
- (a)
- 4.
ADH radioimmunoassay(usually measured at the end of dehydration test): Levels are low in central DI and high in nephrogenic DI; then continue with:
- 5.
Vasopressin test:
- (a)
Once the diagnosis of DI is established, the ADH-insensitive (nephrogenic) disease must be distinguished from the ADH-sensitive (central) form; this is done by giving five units of vasopressin parenterally or 2 μg of desmopressin and then measuring urinary flow and D for two more hours:
In central DI: urinary flow ↓ and urinary density and osmolality ↑
In nephrogenic DI: urinary flow remains ↑ and urinary density and osmolality ↓
- (a)
Investigation of the Cause
MRI head
Looking for tumors (e.g., hypothalamic, pineal, or infiltration).
May demonstrate loss of bright spot of the posterior pituitary gland.
Abdominal ultrasound, intravenous urography: for the renal pathology.
Differential Diagnosis of Polyuria–Polydipsia Syndrome
Primary polydipsia
It involves greatly increased drinking in excess of 5 l/day, leading to dilution of the extracellular fluid, inhibition of ADH secretion, and water diuresis (there may be impaired renal effectiveness because of washout of solute and, therefore, reduced urine-concentrating ability).
It is a disorder of thirst that is either due to psychogenic causes or to altered osmotic and nonosmotic regulation of thirst.
Other causes of polyuria: renal chronic failure (polyuric phase), DM, hypercalcemia, hypokalemia
3.3.1.5 Treatment
Central Diabetes Insipidus
Hormonal treatment of substitution: Desmopressin acetate
Vasopressin analog, acting predominantly on the V2 receptors in the kidney.
The drug may be administered in divided doses, orally—Minirin® (0.2–1.2 mg/day), intranasally (10–40 μg/day), parenterally (SC, IV, IM) (0.1–2 μg/day) or sublingually.—Minirin melt® 60–120 μg: three times/day.
Monitoring of serum sodium and osmolality is essential, as hyponatremia or hypo-osmolality may develop.
Etiologic treatment: the underlying disorder should be treated if possible.
Nephrogenic Diabetes Insipidus
Correction of the underlying cause (metabolic or drugs).
High doses of desmopressin (e.g., up to 5 μg IM) can be effective.
Maintenance of adequate fluid input.
Thiazide diuretics—Hydrochlorothiazide (NEFRIX) 25–50 mg/day + low sodium diet.
3.4 Acromegaly and Gigantism
GH-secreting pituitary adenomas are second in frequency to prolactinomas and cause the classic clinical syndromes of acromegaly and gigantism.
The characteristic clinical features are the consequence of chronic excessive GH and hence IGF-1 secretion. The secretion of GH is autonomic, persistent and the GH’s dynamic control is abnormal. Overgrowth of bone is the classic feature, but GH excess causes a generalized systemic disorder with deleterious effects and an increased mortality rate, although deaths are rarely due to the space-occupying or destructive effects of pituitary adenoma.
In adults, GH excess leads to acromegaly, the syndrome characterized by local overgrowth of bone, particularly of the skull and mandible. Linear growth does not occur because of prior fusion of the epiphyses of long bones.
In childhood and adolescence (prior fusion of the epiphyses), the onset of chronic GH excess leads to gigantism. Most of these patients have associated hypogonadism, which delays epiphyseal closure and the combination of IGF-I excess and hypogonadism leads to a striking acceleration of linear growth (height >+3 SD for age and sex). Most patients with gigantism also have features of acromegaly if GH hypersecretion persists through adolescence and into adulthood.
GH and IGF-1 effects and pathophysiology of acromegaly
- 1.
Growth promoting
- (a)
Most of the deleterious effects of chronic GH hypersecretion are caused by its stimulation of excessive amounts of IGF-1. IGF-1 has growth-promoting effects (DNA, RNA, and protein synthesis) → to the characteristic proliferation of bone, cartilage, and soft tissues and ↑ in size of other organs.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


- (a)