Pharmacokinetics and Pharmacodynamics of Anticancer Drugs
Alex Sparreboom
Sharyn D. Baker
INTRODUCTION
Drug selection and therapy considerations in oncology were originally solely based on observations of the effects produced.1 To overcome some of the limitations of this empirical approach and to answer questions related to considerations of dose, frequency, and duration of drug treatment, it is necessary to understand the events that follow drug administration. Preclinical in vitro and in vivo studies have shown that the magnitude of antitumor response is a function of the concentration of drug,2 and this has led to the suggestion that the therapeutic objective can be achieved by maintaining an adequate concentration at the site of action for the duration of therapy.3 However, drugs are rarely directly administered at their sites of action. Indeed, most anticancer drugs are given intravenously or orally, and yet are expected to act in the brain, lungs, or elsewhere. Drugs must, therefore, move from the site of administration to the site of action and, moreover, distribute to all other tissues including organs that eliminate them from the body, such as the kidneys and liver. To administer drugs optimally, knowledge is needed not only of the mechanisms of drug absorption, distribution, and elimination, but also of the kinetics of these processes.4
The treatment of human malignancies involving drugs can be divided into two pharmacologic phases, a pharmacokinetic phase in which the dose, dosage form, frequency, and route of administration are related to drug level-time relationships in the body, and a pharmacodynamic phase in which the concentration of drug at the site(s) of action is related to the magnitude of the effect(s) produced. Once both of these phases have been defined, a dosage regimen can be designed to achieve the therapeutic objective, although additional factors need to be taken into consideration (Fig. 15.1). The clinical application of this approach allows distinctions between pharmacokinetic and pharmacodynamic causes of an unusual drug response. A basic tenet of pharmacokinetics is that the magnitude of both the desired response and toxicity are functions of the drug concentration at the site(s) of action. Accordingly, therapeutic failure results when either the concentration is too low, resulting in ineffective therapy, or is too high, producing unacceptable toxicity. Between these limits of concentrations lies a region associated with therapeutic success, the so-called therapeutic window.5 Because the concentration of a drug at the site of action can rarely be measured directly, with the exception of certain hematologic malignancies, plasma or blood is commonly measured instead as a more accessible alternative.
PHARMACOKINETIC CONCEPTS
A drug’s pharmacokinetic properties can be defined by two fundamental processes affecting drug behavior over time, absorption and disposition.
Absorption
Historically, most anticancer drugs have been administered intravenously; however, the use of orally administered agents is growing with the development of small-molecule targeted cancer therapeutics, such as tyrosine kinase inhibitors.6 Moreover, drugs may also be administered regionally, for example into the pleural or peritoneal cavities,7 the cerebrospinal fluid, or intra-arterially into a vessel leading to a cancerous tissue.8 The process by which the unchanged drug moves from the site of administration to the site of measurement within the body is referred to as absorption. Loss at any site prior to the site of measurement contributes to a decrease in the apparent absorption of a drug. For an orally administered agent, this complex series of events involves disintegration of the pharmaceutical dosage form, dissolution, diffusion through gastrointestinal fluids, permeation of the gut membrane, portal circulation uptake, passage through the liver, and, finally, entry into the systemic circulation. The loss of drug as it passes for the first time through organs of elimination, such as the gastrointestinal membranes and the liver, during the absorption process is known as the first-pass effect.9
The pharmacokinetic parameter most closely associated with absorption is availability or bioavailability (F), defined as the fraction (or percent) of the administered dose that is absorbed intact. Bioavailability can be estimated by dividing the area under the plasma concentration-time curve (AUC) achieved following extravascular administration by the AUC observed after intravenous administration, and can range from 0 to 1.0 (or 0% to 100%).
Disposition
Disposition is defined as all the processes that occur subsequent to absorption of a drug; by definition, the components of disposition are distribution and elimination. Distribution is the process of reversible transfer of a drug to and from the site of measurement. Any drug that leaves the site of measurement and does not return has undergone elimination, which occurs by two processes, excretion and metabolism. Excretion is the irreversible loss of the chemically unchanged drug, whereas metabolism is the conversion of drug to another chemical species.
The extent of drug distribution can be determined by relating the concentration obtained with a known amount of drug in the body and is, in essence, a dilution space. The apparent volume into which a drug distributes in the body at equilibrium in called the volume of distribution (Vd), and may or may not correspond to an actual physiologic compartment.
The rate and extent to which a drug distributes into various tissues depend on a number of factors, including hydrophobicity, tissue permeability, tissue-binding constants, binding to serum proteins, and local organ blood flow.10 Large apparent volumes of distribution are common for agents with high tissue binding or high lipid solubility,
although distribution into specific body compartments may be limited by physiologic processes, such as the blood-brain barrier protecting the central nervous system11,12 or the blood-testes barrier.13
although distribution into specific body compartments may be limited by physiologic processes, such as the blood-brain barrier protecting the central nervous system11,12 or the blood-testes barrier.13
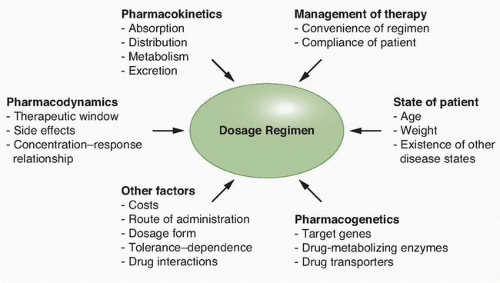 Figure 15.1 Principal determinants of dosage regimen selection for an anticancer drug |
Just as Vd is needed as a parameter to relate the concentration to the amount of drug in the body, there is also a need to have a parameter to relate the concentration to the rate of drug elimination, which is known as clearance (CL). Of all pharmacokinetic parameters, CL has the most clinical relevance because it defines the key relationship between drug dose and systemic drug exposure (AUC). Derived from Vd and CL is the parameter elimination rate constant, which can be regarded as the fractional rate of drug removal. It is, however, more common to refer to the half-life than to the elimination rate constant of a drug. The half-life of a drug is a useful parameter to estimate the time required to reach steady state on a multidose schedule or during a continuous intravenous drug infusion.
Dose Proportionality
When drug concentrations change in strict proportionality to the dose of drug administered, then the condition of dose proportionality (or linear pharmacokinetics) holds. If doubling the dose exactly doubles the plasma concentration or AUC, then pharmacokinetic parameters such Vd, and CL are constant and remain independent of dose and concentration.14 By strict definition, drugs with linear pharmacokinetics are dose proportional. Dose proportionality is clinically important because it means that dose adjustments will generate predictable changes in systemic drug exposure. For drugs that lack dose proportionality, Vd and CL will demonstrate concentration or time dependence, or both, making it difficult to predict the effect of dose adjustments on drug concentration (Fig. 15.2). Factors that can contribute to a lack of dose proportional pharmacokinetics include saturable oral absorption,15 capacity-limited distribution or protein binding,16 and/or saturable metabolism.17 Dose proportionality of anticancer agents is typically assessed in Phase 1 dose-escalation trials in which small groups of patients are treated at a single dose level using a parallel study design, although the statistical power of such studies to detect deviations from dose proportionality is poor. An alternative, more robust study design is a crossover study in which each patient receives a low dose, an intermediate dose, and a high dose over consecutive cycles of treatment.18 However, such studies are relatively rare in oncology because of the required use of low, potentially ineffective doses, which may raise ethical concerns for patients.
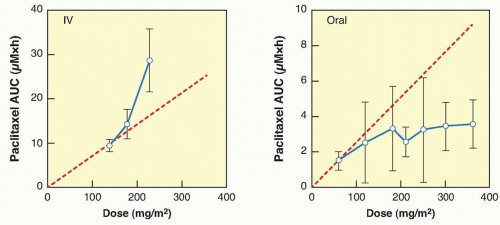 Figure 15.2 Effect of drug dose on systemic exposure to paclitaxel following intravenous (IV) or oral administration in patients with cancer. Data are expressed as mean values (symbols) and standard deviation (error bars). The dashed line indicates the hypothetical dose-proportional increase in the area under the plasma concentration time curve (AUC). (Data derived from van Zuylen L, Karlsson MO, Verweij J, et al. Pharmacokinetic modeling of paclitaxel encapsulation in Cremophor EL micelles. Cancer Chemother Pharmacol 2001;47:309-318, and Malingre MM, Terwogt JM, Beijnen JH, et al. Phase I and pharmacokinetic study of oral paclitaxel. J Clin Oncol 2000;18:2468-2475, respectively.) |
TABLE 15.1 Examples of Systemic Exposure as a Pharmacodynamic Marker of Anticancer Drug Effects | |||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
PHARMACODYNAMIC CONCEPTS
Pharmacodynamic models relate clinical drug effects with drug dose, concentration, or other pharmacokinetic parameters indicative of drug exposures (Table 15.1). In oncology, pharmacodynamic variability may account for substantial differences in clinical outcomes, even when systemic exposures are uniform. Variability in pharmacodynamic response may be heavily influenced by clinical covariates such as age, gender, prior chemotherapy, prior radiotherapy, concomitant medications, or other variables.19 The pharmacokinetic parameters that are most often correlated with drug effects are markers of drug exposure, such as AUC. In general, the specific parameter used as the independent variable in a pharmacodynamic analysis depends on the particular characteristics of the study drug.
In oncology, pharmacodynamic studies of drug effects have most often focused on toxicity endpoints.20 Continuous response variables, such as the percentage fall in the absolute blood count from baseline, are easily analyzed using nonlinear regression methods. Dose-limiting neutropenia has been frequently analyzed using a sigmoid maximum effect model described by the modified Hill equation. The pharmacodynamic analysis of subjectively graded clinical endpoints, such as common toxicity criteria scores on a 4-point scale, may require more sophisticated statistical methods.21,22 Logistical regression methods have been used to model these types of categorical (ordinal) response or outcome variables.
Physiologic pharmacodynamic models describing the severity and time course of drug-related myelosuppression have been derived using population mixed-effect methods for several agents, including paclitaxel23,24 and pemetrexed.25 The ability of these models to predict both the severity and duration of drug-induced neutropenia substantially enhances their clinical usefulness.26 In contrast to small-molecule therapeutics, large-molecule therapeutics such as monoclonal antibodies may not demonstrate toxicities directly related to dose levels. For these agents, a thorough understanding of the pharmacokinetic/pharmacodynamic relationships using modeling approaches may be critical for optimal dose selection.27
The antitumor activity of certain chemotherapeutic agents is highly schedule dependent. For such drugs, the dose fractionated over several days can produce a different antitumor response or toxicity profile compared with the same dose given over a shorter period. For example, the efficacy of etoposide in the treatment of small-cell lung cancer is markedly increased when an identical total dose of etoposide is administered by a 5-day divided-dose schedule rather than a 24-hour infusion.28 Pharmacokinetic analysis in that study showed that both schedules produced very similar overall drug exposure (as measured by AUC), but that the divided-dose schedule produced twice the duration of exposure to an etoposide plasma concentration of >1 µg/mL. This finding has led to the use of prolonged oral administration of etoposide to treat patients with cancer.29 Similar schedule dependence has been demonstrated for a number of other anticancer agents, notably paclitaxel30,31 and topotecan.32 For these agents, the variability in clinically tested treatment schedules is enormous, ranging from short intravenous infusions of less than 30 minutes to 21-day or even 7-week continuous infusion administrations, with large differences in experienced toxicity profiles.
VARIABILITY IN PHARMACOKINETICS/PHARMACODYNAMICS
There is often a marked variation in drug handling between individual patients, resulting in variability in pharmacokinetic parameters (Fig. 15.3), which will often lead to variability in the pharmacodynamic effects of a given dose of a drug.33 That is, an identical dose of drug may result in acceptable toxicity in one patient, and unacceptable and possibly life-threatening toxicity in another, or a clinical response in one individual and cancer progression in another. The principal underlying sources of this interindividual pharmacokinetic/pharmacodynamic variability are discussed in the following paragraphs.
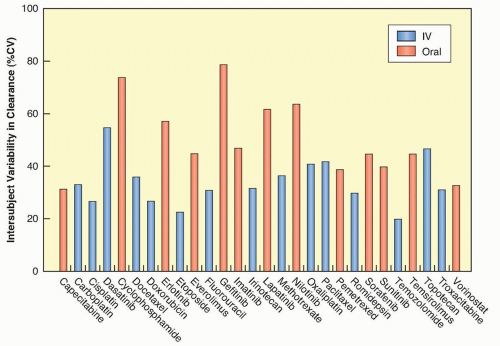 Figure 15.3 Interindividual pharmacokinetic variability of select cytotoxic agents and molecularly targeted agents expressed as a percent coefficient of variation (%CV) in apparent (oral) clearance. IV, intravenous. (Data derived from Mathijssen RH, de Jong FA, Loos WJ, et al. Flat-fixed dosing versus body surface area based dosing of anticancer drugs in adults: does it make a difference? Oncologist 2007;12:913-923, and publicly available prescribing information.) |
Body Size and Body Composition
The traditional method of individualizing anticancer drug dosage is by using body surface area (BSA).34 However, the usefulness of normalizing an anticancer drug dose to BSA in adults has been questioned, because, for many drugs, there is no relationship between BSA and CL.35 Likewise, attempts to replace BSA as a size metric in dose calculation with alternate descriptors such as lean body weight, either in an average population or in individuals at the outer extremes of weight (i.e., frail, severely obese patients) have failed for many anticancer agents.36,37 It should be pointed out that BSA is a much more important consideration in drug dose calculation for pediatric patients as compared to adults, because of the larger size range in the former population.38 Based in part on the failure to reduce interindividual pharmacokinetic variability with the use of BSA normalization to obtain a starting dose, many of the more recently developed molecularly targeted agents are currently administered using a flat-fixed dose irrespective of an individual’s BSA.37
Age
Changes in body composition and organ function at the extremes of age can affect both drug disposition and drug effect.39 For example, maturational processes in infancy may alter the absorption and distribution of drugs as well as change the capacity for drug metabolism and excretion.4 The importance of understanding the influence of age on the pharmacokinetics and pharmacodynamics of individual anticancer agents has increased steadily as treatment for the malignancies of infants,40 adolescents,41 and the elderly42 has advanced. Although pediatric cancers remain rare compared with cancers in adults and the elderly population, in particular, optimizing treatment in a patient group with a high cure rate and a long expected survival becomes critical to minimize the incidence of preventable late complications while maintaining efficacy.
Pathophysiologic Changes
Effects of Disease
Pathophysiologic changes associated with particular malignancies may cause dramatic alterations in drug disposition. For example, increases in the clearance of both antipyrine and lorazepam were noted after remission induction compared with the time of diagnosis in children with acute lymphoblastic leukemia (ALL).43 The clearance of unbound teniposide is lower in children with ALL in relapse than during first remission.44 Because leukemic infiltration of the liver at the time of diagnosis is common, drugs metabolized by the liver may have a reduced clearance, as has been documented in preclinical models.45
Furthermore, in mouse models, certain tumors elicited an acute phase response that coincided with downregulation of human CYP3A4 in the liver as well as the mouse ortholog Cyp3a11.46 The reduction of murine hepatic Cyp3a gene expression in tumor-bearing mice resulted in decreased Cyp3a protein expression and, consequently, a significant reduction in Cyp3a-mediated metabolism of midazolam. These findings support the possibility that tumor-derived inflammation may alter the pharmacokinetic and pharmacodynamic properties of CYP3A4 substrates, leading to reduced metabolism of drugs in humans.47 This supports a possible need for disease-specific design of early clinical trials with anticancer drugs,48 as has been recommended for docetaxel.49
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


