Platinum Analogs
Peter J. O’Dwyer
A. Hilary Calvert
INTRODUCTION
The platinum drugs represent a unique and important class of antitumor compounds. Alone or in combination with other chemotherapeutic agents, cis-diamminedichloroplatinum (II) (cisplatin) and its analogs have made a significant impact on the treatment of a variety of solid tumors for nearly 40 years. The unique activity and toxicity profile observed with cisplatin in early clinical trials fueled the development of platinum analogs that are less toxic and more active against a variety of tumor types, including those that have developed resistance to cisplatin. In addition to cisplatin, two other platinum complexes are currently approved for use in the United States: cis-diamminecyclobutanedicarboxylate platinum (II) (carboplatin) and 1,2-diaminocyclohexaneoxalato platinum (II) (oxaliplatin). Several other analogs with unique activities are in various stages of clinical development, and nedaplatin (Japan) and lobaplatin (China) are locally registered. Progress in the development of superior analogs requires a thorough understanding of the chemical, biologic, pharmacokinetic, and pharmacodynamic properties of this important class of drugs.
HISTORY
The realization that platinum complexes exhibited antitumor activity began serendipitously in a series of experiments to investigate the effect of electromagnetic radiation on the growth of bacteria, carried out by Dr. Barnett Rosenberg and colleagues beginning in 1961.1,2 Exposure of the bacteria to an electric field resulted in a profound change in their morphology; this effect was found not to be from the electric field, but from electrolysis products produced by the platinum electrodes. An analysis of these products resulted in the identification of the cis-isomer of a platinum coordination complex as the active compound. Tests of cis-diamminedichloroplatinum (II) in mice bearing several model tumor types indicated that cisplatin exhibited a broad spectrum of antitumor activity. Although early clinical trials demonstrated responses in several tumor types, particularly testicular cancers, the severe renal and gastrointestinal toxicity caused by the drug nearly led to its abandonment. Work at Memorial Sloan-Kettering3,4 showed that these effects could be ameliorated, in part, by aggressive prehydration, which rekindled interest in its clinical use. Currently, cisplatin is curative in testicular cancer and significantly prolongs survival in combination regimens for ovarian, lung, head and neck, bladder, and upper gastrointestinal (GI) cancers. Its role is being reexamined in other tumors, too, and especially breast cancer.
PLATINUM CHEMISTRY
Platinum exists primarily in either a 2+ or 4+ oxidation state. These oxidation states dictate the stereochemistry of the ligands surrounding the platinum atom. Platinum (II) compounds exhibit a square planar geometry, in which the ammine ligands (also called carrier groups) are relatively stable, whereas the opposite, more polar ligands (leaving groups) are more easily displaced and so confer reactivity toward charged macromolecules, including DNA.5 The stereochemistry of platinum complexes is critical to their antitumor activity as evidenced by the significantly reduced efficacy observed with trans-diamminedichloroplatinum (II).
In an aqueous solution, the chloride leaving groups of cisplatin are subject to mono- and diaqua substitution, particularly at chloride concentrations below 100 mmol, which characterize the intracellular environment. The administration of cisplatin in high chloride solutions (normal saline usually), therefore, contributes to stability. Intracellular formation of partially and fully aquated complexes creates the chloroaqua and hydroxoaqua cisplatin species that bind DNA.6
PLATINUM COMPLEXES AFTER CISPLATIN
Early in the clinical development of cisplatin, it became clear that its toxicity was a limitation to its therapeutic effectiveness, and that its activity, although striking in certain diseases, did not extend to all cancers. These observations then motivated a search for structural analogs with less toxicity and a different profile of antitumor activity. In addition, the side effects of cisplatin stimulated the development of antiemetics and other supportive care measures for use with chemotherapy. Progress in understanding the chemistry and pharmacokinetics of cisplatin has guided the development of new analogs. In general, modification of the chloride leaving groups of cisplatin results in compounds with different pharmacokinetics and reactivity towards DNA, whereas modification of the carrier ligands alters the activity of the resulting complex. The features of the more important platinum analogs that have been developed are shown in Figure 18.1.
Carboplatin
The carboplatin molecule has the same ammine carrier ligands as cisplatin. Using a murine screen for nephrotoxicity, Harrap and Calvert discovered that substituting a cyclobutanedicarboxylate moiety for the two chloride ligands of cisplatin resulted in a complex with reduced renal toxicity. This observation was translated to the clinic in the form of carboplatin, a more stable and pharmacokinetically predictable analog.7,8 The results in humans were accurately predicted by the animal models, and marrow toxicity rather than nephrotoxicity was the principal side effect. At effective doses, carboplatin produced less nausea, vomiting, nephrotoxicity, and neurotoxicity than cisplatin. Furthermore, the myelosuppression was closely associated with the pharmacokinetics. The work of Calvert et al.9 and Egorin and colleagues10 showed that toxicity can be made more predictable and dose intensity less variable by dosing strategies based on the exposure. Carboplatin was shown to be indistinguishable from cisplatin in its clinical activity in all but a handful of tumor types and is the most frequently used form of platinum in current use. Cisplatin and carboplatin have almost superimposable profiles of activity in the NCI60 cell line screen, which further emphasizes the dependence of spectrum of activity on the carrier ligand.
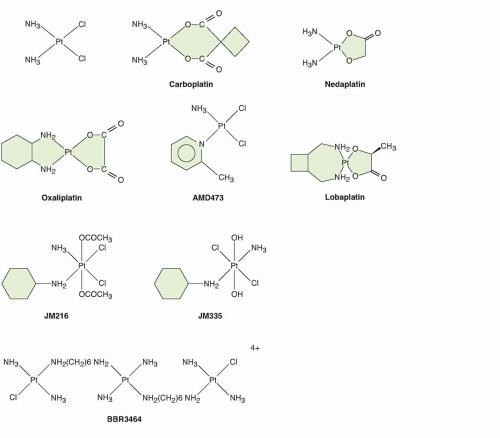 Figure 18.1 Structures of cisplatin, analogs, lobaplatin, and nedaplatin. |
Oxaliplatin
Compounds with activity in cisplatin-resistant models emerged from modifications to the carrier group (see left side of the analogs in Fig. 18.1). Connors, in the late 1960s, synthesized platinum coordination compounds with varying physicochemical characteristics and found that the series that possessed a diaminocyclohexane (DACH) carrier group was active in models of cancer in vitro11 and in vivo.12 Subsequent studies supported the idea that DACH-based platinum complexes were non-cross-resistant with cisplatin, and DACH derivatives exhibited a unique cytotoxicity profile compared to cisplatin and carboplatin in the National Cancer Institute 60 cell line screen.13,14,15 After a number of delays, a DACH analog that had been synthesized by Kidani and colleagues in the early 1970s, was developed in the clinic.13 Oxaliplatin, a coordination compound of a DACH carrier group and an oxalato leaving group, was active in cisplatin-resistant tumor models. Like cisplatin, oxaliplatin preferentially forms adducts at the N7 position of guanine and, to a lesser extent, adenine. However, there is evidence that the three-dimensional structure of the DNA adducts and biologic response(s) they elicit are different from those of cisplatin. Oxaliplatin demonstrated activity in combination with 5-fluorouracil and leucovorin in colon cancer, a disease that is unresponsive to cisplatin. This finding validated the focus on cisplatin-resistant preclinical models to identify new active molecules. Oxaliplatin is approved for the treatment of advanced colorectal cancer, and enhances cure rates in the adjuvant setting. The therapeutic role of oxaliplatin has been found to extend to pancreatic, gastric, and esophageal cancers, in all of which it is the more active platinum derivative.
Nedaplatin and Lobaplatin
Nedaplatin is cis-diammineglycolatoplatinum, developed as a less nephrotoxic second-generation platinum analog, has been shown to be active in a range of tumors similar to that of cisplatin and carboplatin.16 As a diammine structure, nedaplatin would fall among the cisplatin analogs analyzed in the NCI60 cell line screen,17 and this activity is therefore anticipated. Lobaplatin is a platinum (II) complex in which the leaving group is lactic acid and the stable ammine ligand is 1,2-bis(aminomethyl)cyclobutane. In a similar way to oxaliplatin the stable ammine ligand may convey some non-cross-resistance compared to cisplatin or carboplatin. It is licensed in China for the breast cancer, small-cell lung cancer, and chronic myelogenous leukemia. It is unique among the platinum drugs for its approval for breast cancer, but there are few published clinical data and no randomized trials. It has not achieved approval in the United States or Europe.
Newer Platinum Structures
The octahedral stereochemistry adopted by platinum (IV) compounds has led investigators to speculate that they may exhibit a different spectrum of activity than that of platinum (II) drugs. Two compounds that were tested clinically without much success are ormaplatin and iproplatin. Two other platinum (IV) compounds that exhibit novel structural features, satraplatin (previously JM216) and JM335 (trans-ammine[cyclohexylamine]dichlorodihydroxo platinum [IV]), underwent more limited development. Satraplatin was the first orally active platinum compound, and showed some activity in lung and ovarian cancers, but despite promising activity in prostate cancer, a phase III trial was not successful.18,19
An approach based on the chemistry of the platinum-DNA interaction led to design and synthesis by Farrell et al.20 of a novel class of compounds containing multiple platinum atoms (see Fig. 18.1). These bi- and trinuclear structures form adducts that span greater distances across the minor groove of DNA and have a profile of cell kill that differs from that of the small molecules. These compounds are unique in that their interaction with DNA is considerably different from that of cisplatin, particularly in the abundance of interstrand cross-links formed. Clinical development of candidate compounds is at a preliminary stage.
Efforts have been made to design novel platinum analogs that can circumvent putative cisplatin resistance mechanisms. An example is cis-amminedichloro(2-methylpyridine) platinum (II) (also known as AMD473 and ZD0473). This compound is a sterically hindered platinum complex that was designed to have minimal reactivity with thiols and thus avoid inactivation by molecules such as glutathione.21,22 Responses were identified with its use in the clinic, but development was curtailed based on low levels of activity. The recent description of a monofunctional platinum (II) analog, phenanthriplatin, from the lab of Lippard is potentially of great interest, based on both potency in vitro and a mechanistic profile different from existing analogs.23 A renewed appreciation that chemotherapeutic drugs have a continuing role in managing cancer is likely to prompt additional clinical development of novel platinum structures.
MECHANISM OF ACTION
DNA Adduct Formation
DNA has long been thought to be the major therapeutic target for platinum compounds. The cytotoxic effects are determined, in part, by the structure and relative amount of DNA adducts formed. Cisplatin and its analogs react preferentially at the N7 position of guanine and adenine residues to form a variety of monofunctional and bifunctional adducts.24 The monoadducts may form intrastrand or interstrand cross-links. The predominant lesions that are formed when platinum compounds bind DNA are d(GpG)Pt intrastrand cross-links. Cisplatin also forms interstrand cross-links between guanine residues located on opposite strands, and these account for less than 5% of the total DNA-bound platinum. The formation of adducts and cross-links has been associated with therapeutic efficacy.25,26 These adducts may contribute to the drug’s cytotoxicity because they impede certain cellular processes that require the separation of both DNA strands, such as replication and transcription. The adducts formed in the reaction between carboplatin and DNA in cultured cells are essentially the same as those of cisplatin; however, higher concentrations of carboplatin are required (20- to 40-fold for cells) to obtain equivalent total platinum-DNA adduct levels due to its slower rate of aquation.27 Oxaliplatin intrastrand adducts form even more slowly due to a slower rate of conversion from monoadducts; however, they are formed at similar DNA sequences and regions as cisplatin adducts. At equitoxic doses, oxaliplatin forms fewer DNA adducts than does cisplatin. This has been interpreted to mean that oxaliplatin lesions are more cytotoxic than those formed by cisplatin.
The differences observed in cytotoxicity between the diammine (e.g., cisplatin, carboplatin) and DACH platinum compounds may not depend on the type and relative amounts of the adducts formed, but on the overall three-dimensional structure of the adduct and its recognition by various cellular proteins. The major difference between them is the protrusion of the DACH moiety of oxaliplatin into the major groove of DNA, which thus produces a bulkier adduct than that of cisplatin. This bulkier, more hydrophobic adduct seems to be recognized differently by cellular proteins involved in sensing DNA damage.28 The functional consequences are twofold: Proteins such as polymerases that recognize and participate in reactions on DNA under normal circumstances may be perturbed, whereas processes that are controlled by proteins that recognize damaged DNA may become activated (the DNA damage response). The latter group of proteins function both in the DNA repair process and in cellular signaling toward cell survival/death decisions.
DNA Interstrand Cross-Links
Although the DNA adducts are well-recognized to result in G-G interstrand cross-links, like classical alkylating agents, platinum drugs have the capacity to form intrastrand cross-links, albeit to a lesser degree. By blocking essential aspects of DNA metabolism, such as replication and transcription, intrastrand cross-links are highly cytotoxic. Recent studies have drawn attention both to the cytotoxicity of these lesions, and their differing mechanisms of repair, both replication dependent and independent.29,30 These studies may have clinical implications in selecting patients for therapy based on the repair competence of tumors.
CELLULAR RESPONSES TO PLATINUM-INDUCED DNA DAMAGE
Multiple cellular outcomes may follow the formation of platinum-DNA adducts, including cell death by apoptosis, necrosis, or mitotic catastrophe, or cell survival by activation of various protective mechanisms including DNA repair, DNA damage signaling pathways, cell cycle arrest, and autophagy (the last may have a dual role, possibly context dependent).
Cell Fate
The cellular effects following DNA binding by platinum drugs have been analyzed. The studies of Sorenson and Eastman,31 using DNA repair-deficient Chinese hamster ovary (CHO) cells, indicated that passage through the S phase is necessary for G2 arrest and cell death, which suggests that DNA replication on a damaged template may result in the accumulation of further damage. An aberrant mitosis was observed before apoptosis in this model.
DNA Damage Recognition
Among the initiation events that ultimately result in platinum drug-induced cell death are the binding of platinum-DNA damage recognition proteins, which then seed the accumulation of a large protein complex capable both of DNA damage signaling (as to cell cycle proteins to halt replication) and repair of the damaged DNA. Among the DNA-binding proteins are the high-mobility group proteins HMG1 and HMG2.32,33,34 These proteins are capable of bending DNA as well as recognizing bent DNA structures, such as that produced by cisplatin, and different specificities for cisplatin and for oxaliplatin adducts are observed in structural studies.35,36 Other candidate platinum-DNA damage recognition proteins include histone H1, RNA polymerase I transcription upstream binding factor (hUBF), the TATA binding protein (TBP), and proteins
involved in mismatch repair (MMR). The MMR complex has been implicated in cisplatin sensitivity.37 Studies have shown that the MSH2 and MLH1 proteins participate in the recognition of DNA adducts formed by cisplatin, but not oxaliplatin, which could contribute to differences in the cytotoxicity profiles observed between these two platinum complexes.
involved in mismatch repair (MMR). The MMR complex has been implicated in cisplatin sensitivity.37 Studies have shown that the MSH2 and MLH1 proteins participate in the recognition of DNA adducts formed by cisplatin, but not oxaliplatin, which could contribute to differences in the cytotoxicity profiles observed between these two platinum complexes.
DNA Damage Signaling
A number of signaling events have been shown to occur after treatment of cells with platinum drugs.38 For example, the ATM- and Rad3-related (ATR) proteins that are involved in cell-cycle checkpoint activation are activated by cisplatin. These kinases phosphorylate and activate several downstream effectors that regulate cell cycle, DNA repair, cell survival, and apoptosis, including p53, CHK2, and members of the mitogen-activated protein kinase (MAPK) pathway (extracellular signal-related kinase [ERK], c-Jun amino-terminal kinase [JNK], and p38 kinase). Recent data especially implicate signaling through the JNK pathway, and inhibition at the level of JNK seems especially relevant to platinum drug cytotoxicity in vitro and in vivo.39,40 The pleiotropic nature of this stress response only grows, because each of these molecules subsequently controls the activity and expression of many more proteins. As a result of this complexity, acting in the context of variable genomic tumor aberrations, therapeutic strategies directed to these pathways have been slow to emerge. However, clinical trials to investigate specific inhibitors of DNA damage responses are underway and hold promise. It is also relevant to point out that these signaling pathways affect not just the tumor cell, but also may communicate to cells in the microenvironment, the responses of which may also determine the effectiveness of therapy.
IS DNA THE ONLY TARGET?
Early analyses of the action of cytotoxic drugs included a probe of whether effects on DNA were sufficient to explain drug effects. A pioneer in this field was Tritton,41 who proposed that effects of DNA-intercalating agents on the plasma membrane could underlie the cytotoxicity of the drug. More recently, enucleated cells were shown to be susceptible to cisplatin, and a seminal paper from Voest and colleagues showed that platinum sensitivity was determined not solely by the accumulation of DNA damage in the tumor cell.42 In analyzing the contribution of cells in the microenvironment of tumors, he showed that tumor infiltration with mesenchymal stem cells could confer drug resistance. A search for secreted factors defined platinum-induced fatty acids, metabolic products in the thromboxane synthetase, and cyclooxygenase-1 pathways as determining the effectiveness of drug therapy. A proteomic study in cisplatin-sensitive and -resistant cells confirmed the substantial effects of drug exposure on lipid metabolites and their relation to susceptibility. A current focus on therapies directed to the microenvironment, including immunologic and anti-inflammatory interventions,43 has the potential to expand our ability to apply platinum drugs in the clinic.
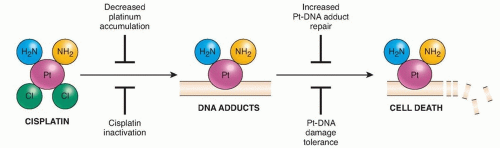 Figure 18.2 Cellular mechanisms of cisplatin resistance. |
MECHANISMS OF RESISTANCE
The major limitation to the successful treatment of solid tumors with platinum-based chemotherapy is the emergence of drug-resistant tumor cells.44 Developments in tumor biology have advanced our thinking with regard to how and when these cells emerge; heterogeneity within a tumor even at its earliest diagnosis reflects the emergence of treatment-resistant clones even in advance of selection pressure and the realization that resistance may not be specific to the DNA-damaging drug. Indeed, this may be reflected clinically in the finding that after progression on initial chemotherapy, the use of second-line therapy is usually associated with a shorter duration of response.
Currently described mechanisms of platinum drug resistance (Fig. 18.2) include reduced cellular accumulation, intracellular detoxification, repair of Pt-DNA lesions, increased damage tolerance, and the activation of cellular defense mechanisms such as autophagy. In addition, we have already alluded to exogenous influences on mechanism, as may be mediated by other cells, metabolites, of physicochemical conditions (such as hypoxia) in the tumor microenvironment. It must be acknowledged, however, that our insights are very limited as to why some tumors respond and others do not to platinum chemotherapy. As genome sequencing yields increasing and often surprising revelations about the genes that drive cancers and the complexity inherent in cancers of a single histologic type, it is likely that when associated with outcomes in large patient populations, patterns will emerge to guide selection of therapies.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


