Candidate Gene Approach
The candidate gene approach to pharmacogenetic investigation attempts to link a phenotype with an alteration in a specific gene through hypothesis-based testing. Recognition that specific genes contribute to variation in drug toxicity occurred in the 1950s with three key discoveries
11 (
Fig. 6-1). Alving et al.
18 identified a relationship between glucose-6-phosphate dehydrogenase deficiency and hemolysis in patients treated with primaquine. Kalow correlated acetylcholinesterase deficiency and prolonged paralysis from succinylcholine.
19 Evans et al.
20 found that the rate of isoniazid acetylation influenced the development of peripheral neuropathy. Since then, variation in numerous other genes has been shown to affect drug efficacy and toxicity, with a 2009 review reporting 541 genes studied.
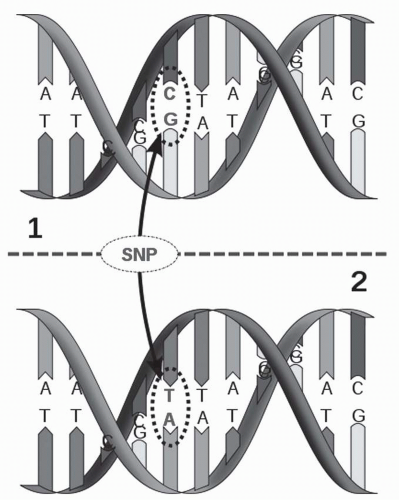
21Even more potential for discovery exists in the over 1.4 million SNPs in the human genome, and 60,000 SNPs in gene-encoding regions.
22 SNPs are common variations that occur at a given location when two DNA sequences are compared,
23 for example:
ATGTA
ATCTA
SNPs occur every 100 to 300 bases and can have a variety of effects—they may be silent (synonymous), producing no change in amino acid sequence, or may change a single amino acid (nonsynonymous) that greatly affects protein function. They may introduce a stop codon, which produces a truncated protein.
24 SNPs that occur in the promoter or enhancer regions of a gene may affect RNA expression and protein levels while SNPs in intron-exon boundaries may lead to splice variants. Within the field of oncology, SNPs
have been associated with numerous variations in responsiveness and side effects of chemotherapeutic agents.
10 The C677T polymorphism in 5,10-methylenetetrahydrofolate reductase (MTHFR) slows the activation of leucovorin rescue and in some studies is associated with increased methotrexate toxicity. Glutathione-S-transferase enzymes metabolize drugs through glutathione conjugation. The GSTP1 Ile105Val mutation is associated with lower enzyme activity and increased survival for oxaliplatin/5-fluorouracil (5-FU) regimens (24.9 months for the V105 homozygote versus 13.3 months for the I105 homozygote).
25,
26 The
Arg399Gln SNP in the DNA repair protein XRCC1 is associated with resistance to oxaliplatin/5-FU (discussed below).
10 Lapatinib, a tyrosine kinase inhibitor metabolized by cytochrome P450 enzymes, is associated with diarrhea and rash in patients with the
CYP2C19*2/*2 SNP.
27 In addition to SNP variants, nucleotide insertions, deletions, repeats, and variations in copy number also contribute to genetic diversity.
28 As technologies to identify SNPs improve, more associations will be added.
Population Studies
Population studies are aimed at the assessment of the frequency of the pharmacogenetic syndrome within the general population as well as frequency differences between populations. As with family studies, population studies can use either phenotypic or genotypic tests. The frequency of individuals with particular phenotypic characteristics can be estimated using the Hardy-Weinberg equation.
31 Population studies to assess the specific phenotype or activity of a particular protein may show unimodal or bimodal patterns of distribution. A unimodal distribution in the activity of a particular enzyme, such as a Gaussian or normal distribution pattern, suggests mutations or genetic alterations that lead to a range of activities in the population studied. In contrast, bimodal distributions may indicate mutations that lead to reduced or greater than normal activity in a subset of the population.
32 The evaluation of particular phenotypes in population studies related to chemotherapeutic drug metabolism may require assessments using a safer probe drug that is metabolized through the same potential polymorphic drug-metabolizing enzyme but without posing a risk of toxicity to the healthy individual participating in the study. Results from a population study comparing cancer patients to healthy volunteers indicated that cancer stage may also influence the phenotype of an important drug-metabolizing enzyme CYP2C19, for which genotype normally predicts phenotype in the healthy population.
13,
33 In this study, patients with advanced cancer had the extensive metabolizer genotype; however, 25% of the patients displayed a poor metabolizer phenotype. This discordance between phenotype and genotype and the decreased activity of CYP2C19 observed in terminally ill cancer patients may influence the clinical efficacy and toxicity of therapeutic agents (e.g., cyclophosphamide) and should be investigated with respect to other drug-metabolizing enzymes.
13 The characterization of genes and related phenotypes involved in anticancer drug pharmacokinetic or pharmacodynamic variability or those involved in cancer predisposition may be limited to investigations using individuals with the particular cancer or those already being treated with a particular chemotherapeutic agent.
After an assessment of phenotypic and genotypic markers in the general population, it may be useful to undertake surveys in other populations, including patients affected with specific types of cancer or being treated with a particular chemotherapeutic agent. Such studies may define the frequency of the pharmacogenetic syndrome in the cancer patient population at risk. Other demographic factors, such as race, age, and gender, may influence the risk to specific groups.
The methodologies discussed above have had specific applications in the identification of SNPs in single genes using the candidate gene approach. The genetic polymorphisms in single genes have not thus far explained a large proportion of the individual variability in drug response, mainly because most drug response phenotypes are thought to be determined by the interactions among multiple genes as well as with the environment. Therefore, the candidate gene strategy has limited value for identifying polygenic determinants of variability in drug response. On the other hand, this approach may offer an advantage for testing “specific” biological or pharmacological hypotheses in a given and often limited clinical sample of patients. Genome-wide studies require larger numbers of subjects to achieve statistical power to identify multiple lowpenetrance genes, a requirement seldom fulfilled in small heterogeneous patient samples.
Genomic Approaches
Efforts using a candidate gene approach were highly successful in identifying genetic causes for severe drug toxicity such as related to glucose-6-phosphate dehydrogenase deficiency.
34 A genome-wide scanning (GWAS) approach is now increasingly used to look for variants that influence the risk of disease and explain differences in drug effect conferred by variants in metabolic enzymes, drug transporters, and drug targets.
34 Evolving technologies such as microarrays, proteomics, and mouse genetics promise to provide many exciting results that influence clinical practice. Commercial gene chips are available to measure entire classes of genes (i.e., cytochrome P450s) or the entire human genome.
35 In recognition of the emerging role of pharmacogenetics in the clinic, the NIH roadmap sponsored creation of the 12-center Pharmacogenetics Research Network.
2 Pharmacogenomic approaches are hampered by the requirement for large sample sizes to generate statistically significant results and by the high numbers of false-positive findings.
28 For example, recent work using the International HapMap, which has genotyped 3.3 million SNPs, predicted the thiopurine methyltransferase phenotype, although 96 genes in this analysis ranked higher.
36Gene expression arrays of tumor provide an alternative approach to SNP arrays and gene variant analysis for predicting response to drugs. Arrays are primarily of value in predicting the need for, and potential success of treatment. Rather than relying on polymorphisms in a single gene, expression patterns of multiple genes or proteins can be assayed to create a “signature” reflective of a drug’s effect. These expression patterns may result from gene variants, but verification of the presence of a variant requires gene sequencing and is usually not done in conjunction with the development of predictive arrays. In contrast to the candidate gene approach, genomic experiments provide an unbiased, hypothesis generating picture of drug effects.
28 Using DNA microarrays on ALL cells, 124 genes were associated with resistance or sensitivity to four chemotherapeutic drugs.
37 A parallel study identified 45 genes differentially expressed in patients resistant to all four drugs.
38 Microarray experiments predict the sensitivity of cancer cells to docetaxel and multidrug regimens and estimate response to treatment.
39,
40,
41 Gene signature analysis has also been incorporated into clinical trials of breast cancer patients, and they seemed to predict pathologic complete response.
42One of the first pharmacogenomic tests available is the Onco
type DX, which is recommended in the 2007 Update of Recommendations for the Use of Tumor Markers in Breast Cancer from the American Society of Clinical Oncology and the National Comprehensive Cancer Network 2008 Clinical Practice Guidelines in Oncology Breast Cancer.
35,
43 Onco
type DX is used to identify which newly diagnosed, node-negative, ER-positive breast cancer patients may benefit from tamoxifen, and may not need adjuvant chemotherapy.
43 The Onco
type DX assay uses RT-PCR to gauge the expression of 21 genes, including those involved in proliferation (Ki67, STK 15, surviving, cyclin B1, MYBL2), invasion (stromelysin 3, cathepsin L2), HER2 (GRB7, HER2), estrogen (ER, PGR, BCL2, SCUBE2), miscellaneous (GSTM1, CD68, BAG1), and five reference genes.
44 RNA extracted from paraffin-embedded tissue is used.
44 Gene expression levels are analyzed by an algorithm that calculates a recurrence score that predicts the likelihood of distant recurrence in tamoxifen-treated patients.
44 The goal of the assay is to identify patients with a good prognosis that may avoid adjuvant chemotherapy.
45The use of the Onco
type DX assay in predicting the likelihood of recurrence in women with node-negative ER-positive breast cancer treated with tamoxifen was validated in a trial of 668 patients from the National Surgical Adjuvant and Bowel clinical trial.
44 The recurrence score was used to cluster patients into low-, intermediate-, and high-risk groups. In the patient population studied, 51%, 22%, and 27% of patients fell in the low-, intermediate-, or high-risk groups, respectively, with a 6.8%, 14.3%, and 30.5% chance of distant recurrence at 10 years, respectively.
44 The Onco
type DX assay provided additional information beyond tumor grade as determined by three pathologists.
44 One limitation of the Onco
type DX assay is that results may be influenced by the tumor block selected.
46Other genomics based tests include MammaPrint (FDA approved 2007), which uses a 70-gene profile to classify tumors from stage I or II breast cancer patients as low or high risk for recurrence
47 and the Rotterdam Signature, a 76-gene profile that does not overlap with MammaPrint or Onco
type DX. Medicare covers Onco
type DX, which has shown to be cost-effective. If used to classify a hypothetical cohort of 100 patients, Onco
type DX was estimated to result in an increase in quality-adjusted survival by 8.6 years and reduce cost by $202,828.
48 A prospective validation of Onco
type DX is underway in a randomized clinical trial (TAILORx).
49In the following section, discussion will be considered for the important SNP variants that influence response to and toxicity of commonly used anticancer drugs. The list of potentially important variants is not all-inclusive; the actual confirmation of an important role for a given variant requires characterization of protein function and large-scale trials, which are often beyond the scope of funding available for such research. Thus, many of the variants of interest have only been assessed through retrospective analysis of toxicity or response patterns in trials not designed for pharmacogenetic study. For most of these variants, understanding of the impact on drug metabolism, transport, or target interaction in human patient populations is incomplete. As will be seen below, genetic variants in drug-metabolizing enzymes are of particular relevance in explaining variable drug responses, especially toxicity. The relationship between polymorphism and toxicity is best established by data demonstrating that the variant affects enzyme activity, that enzyme
activity predicts pharmacokinetic behavior, and that drug levels or AUC predict efficacy or toxicity.