Fig. 1.1
[18F]FDG
Although PET facilities are rapidly increasing worldwide, the only PET radiopharmaceutical currently available for diagnosis is [18F]FDG. In keeping with this notion, [18F]FDG is by far the most widely used radiotracer for clinical purposes, but its application has some shortcomings. Since [18F]FDG is a derivative of glucose, which is taken up by cells via glucose transporters, phosphorylated by hexokinase, and retained in the tissue, its high uptake is not only in tumor cells but also in normal tissues, such as the brain and heart, which have high levels of glucose metabolic activity [12, 13]. Hence, [18F]FDG-PET is not suitable for imaging tumors in these tissues. In addition, because of its high uptake in urine and fast excretion from the bladder, it is difficult to detect tumors in this organ and surrounding tissues using [18F]FDG [5]. Most importantly, because of high uptake in inflamed tissues, it is difficult to distinguish tumor from inflammation using [18F]FDG.
To increase the usefulness of PET and to overcome the disadvantages associated with [18F]FDG, it is important to develop new imaging radiotracers, which use alternative mechanisms for tumor visualization and provide different information to that obtained using [18F]FDG. Moreover, higher tumor specificity than the one provided by [18F]FDG could be achieved using new radiotracers.
In the following sections, the author will introduce the main candidates for post-[18F]FDG tumor imaging radiotracers.
1.3 Amino Acids
The amino acid analog most frequently used as a radiotracer is [11C]methionine ([11C]Met, Fig. 1.2) [14, 15]. [11C]Met is easily synthesized using [11C]methyl iodide or [11C]methyl triflate as the radiolabeling agent. Since the precursor for [11C]Met radiosynthesis is L-homocysteine, the product obtained is only the L-isomer. In the normal brain, where protein metabolism levels are low, PET radiotracers reflecting controlled protein biosynthesis/degradation rates are suitable for the detection of glioma tumors in many PET facilities [16, 17]. However, because the S-[11C]methyl group in the cell is relatively easily transferred into other positions, compared to 11C labeling in other positions, the levels of [11C]Met are insufficient to enable evaluation of protein synthesis ability.
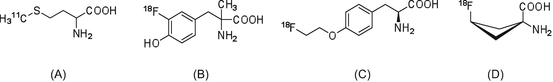
Fig. 1.2
(a) [11C]Met, (b) [18F]FMT, (c) [18F]FET, and (d) [18F]FACBC
Since 1960, amino acids that are stable against metabolism (unlike natural amino acids) have been developed. These artificial amino acids were labeled with 11C and used to detect tumors in preclinical studies. Among them, α-aminoisobutanoic acid, α-aminocyclobutane-1-carboxylic acid, and α-aminocyclopentane-1-carboxylic acid show high binding affinity for amino acid transporters. Moreover, these amino acid analogs do not contain chiral carbon atoms; therefore, their radiolabeled versions are considered promising probes for tumor imaging.
Many 18F-labeled amino acid analogs have been developed and evaluated as candidate of post-[18F]FDG radiotracers (Fig. 1.2). Although phenylalanine and tyrosine analogs were found to correlate with protein synthesis, these analogs are unable to participate in protein synthesis, and their tumor uptake levels are associated with amino acid transporter activity. Then, in efforts to improve in vivo metabolic stability, 3-[18F]fluoro-α-methyl-L-tyrosine ([18F]FMT, Fig. 1.2) [18] and 4-[18F]fluoroethyl-L-tyrosine ([18F]FET) [19] were developed. More recently, [18F]FACBC has been reported as the most promising radiolabeled amino acid analog [20, 21]. This radiotracer has two stereoisomers, with the cis isomer exhibiting higher selectivity than the anti-isomer. In clinical glioblastoma imaging studies, which cannot use [18F]FDG-PET, [18F]FACBC can provide high-quality PET tumor images. Therefore, the 18F-labeled artificial amino acid analogs are viable alternatives for the detection of tumors that cannot be successfully visualized by [18F]FDG-PET.
1.4 Nucleic Acids
[3H]Methylthymidine has been synthesized and used for many years. In addition, methods for production and in vivo evaluation of the PET tracers [11C]methylthymidine (Fig. 1.3) and 2-[11C]thymidine have been reported since 1980 [22, 23]. However, these natural nucleic acid analogs are not stable in vivo, which prompted the modification of their chemical structures and radiolabeling them with 18F, thus creating a probe a longer half-life than 11C.
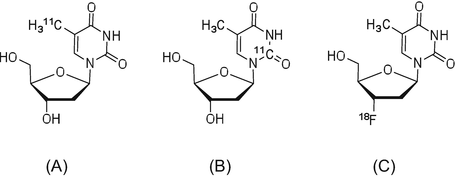
Fig. 1.3
(a) [11C]Methylthymidine, (b) 2-[11C]thymidine, and (c) [18F]FLT
3′-Deoxy-3′-[18F]fluorothymidine ([18F]FLT) is an analog of thymidine, in which 18F is introduced in the 3′-position and shows high in vivo stability and strong resistance to metabolism by cellular thymidine phosphatase [24]. Blood circulating [18F]FLT is taken up by the tissues via a pyrimidine transporter, which functions in nucleic acid synthesis. As the hydroxyl group in 3′-position is replaced by fluorine, phosphorylated [18F]FLT-5′-P cannot participate in the synthesis of DNA, and it is thus retained in the cell as a monophosphate. Hence, the uptake of [18F]FLT can reflect thymidine kinase-1 (TK1) activity in tumor cells [25], which is very low in the G0 stage of the cell cycle and reaches a maximum between the G1 and S phases. Therefore, [18F]FLT has been used to evaluate cell proliferation for tumor staging and assess the therapeutic effects of anticancer drugs.
1.5 Lipid Metabolism
Cancer is characterized by the high proliferation ability of tumor cells, and during this process the synthesis of cell membrane components increases accordingly. Therefore, membrane lipid synthesis is a useful target to also evaluate the proliferation ability of tumor cells. To this end, [11C]acetate, [11C]choline, and [18F]fluorocholine have been developed and used in clinical imaging studies (Fig. 1.4) [26].
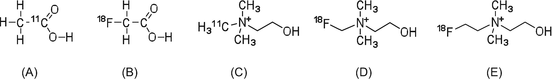
Fig. 1.4
(a) [11C]Acetate, (b) [18F]fluoroacetate, (c) [11C]choline, (d) [18F]fluoromethylcholine, and (e) [18F]fluoroethylcholine
In vitro evaluation of [11C]acetate has been performed to determine the mechanism of radioactivity accumulation in tumor cells [27]. Acetate is formed by the metabolism of phosphatidylcholine and neutral lipids, and, thus, the accumulation of [11C]acetate radioactivity can reflect the proliferation ability of tumor cells [28, 29].
The uptake of choline analogs can reflect the activity of choline kinase and be used to indirectly evaluate ability to synthesize membrane lipids [30–32]. In clinical studies, PET radiotracers for lipid metabolism are useful for the detection of tumors in the brain, the bladder, and the urinary tract. [11C/18F]Choline analogs are not useful for the detection of cancer in epigastrium tissues and other organs because they have a high radioactivity uptake in the liver. Hence, acetate- and choline-based radiotracers may be better to evaluate the therapeutic effects of radiation and antitumor drugs.
1.6 Hypoxia
During the process of tumor cell proliferation, insufficient supply of oxygen results in hypoxia. Hypoxic areas of tumors are relatively insensitive to chemotherapy and radiation therapy. Thus, an understanding of the hypoxic state is useful for the prediction of therapeutic effects and the evaluation of treatment regimens, which has led to the development of PET imaging radiotracers designed to evaluate hypoxia [33–35]. [18F]FMISO (Fig. 1.5) was the first nitroimidazole analog used for imaging of hypoxia in tumors [33]. The nitro group of misonidazole analogs is reduced to form a hydrophilic amine group, and this amine product binds to cellular components and is retained in tumor cells.
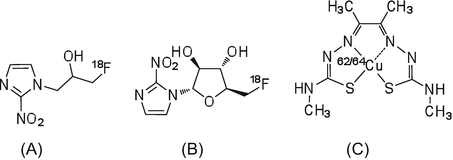
Fig. 1.5
(a) [18F]FMISO, (b) [18F]FAZA, and (c) 62/64Cu-ATSM
In hypoxic areas, which have a low blood flow, the initial uptake of radioactivity by one-pass circulation of radiotracer is low. However, the highly lipophilic [18F]FMISO is able to diffuse into the regions with low blood flow. Unfortunately, the slow clearance of radioactivity from blood means that PET imaging with [18F]FMISO may not result in high-quality images, and thus, extended scanning durations are required [36]. As an alternative, the tracer [18F]FAZA, which has low lipophilicity and high hydrophilicity, has been developed and used in clinical studies [37, 38]. Compared to [18F]FMISO, [18F]FAZA shows improved solubility in water and good signal/noise contrast in PET images within a relatively short PET scanning time.
In addition to nitroimidazole analogs, 62/64Cu-diacetyl-bis(N-4-methylthiosemicarbazone (62/64Cu-ATSM) is also a useful PET imaging radiotracer for hypoxia [39, 40]. Cu-ATSM is a small lipophilic molecular complex that easily penetrates the blood-brain barrier and cellular membranes and clears rapidly from normal tissues [41]. In hypoxic area, Cu2+ binding to ATSM is reduced to Cu+ by microsomal electron transfer, and the Cu+ component is retained in the cell. Compared to [18F]FMISO, 62/64Cu-ATSM shows rapid clearance from normal tissues and blood to produce images with good contrast and signal/noise ratios within a short PET scanning time.
1.7 Receptor and Angiogenesis
By transferring signals through various receptors overexpressed in tumor cells, certain genes and proteins mediate tumor phenotypes, including proliferation ability, invasiveness, metastasis, and treatment resistance. PET studies using radiolabeled tracers for imaging of these receptors are useful for understanding the tumor characteristics.
To date, a large number of PET radiotracers for imaging of receptors have been reported. Major radiotracers of this type include: epidermal growth factor receptor (EGFR) human type (HER2), which is associated with poor prognosis in breast cancer; folic acid receptor, associated with malignant proliferation; chemokine receptor, associated with metastasis; glucagon-like peptide-1 (GLP-1) receptor, associated with neuroendocrine tumors; somatostatin receptor; tumor angiogenesis integrin receptor (αvβ3); and vascular endothelial growth factor (VEGF) [42]. A number of these radiotracers have been used in clinical studies.
1.7.1 Somatostatin Receptors
Somatostatin receptors are G protein-coupled transmembrane proteins that are widely distributed in normal tissues, including those of the central nervous system, pancreas, anterior pituitary, thyroid gland, spleen, gastrointestinal tract, and adrenal gland. There are five somatostatin receptors, of which somatostatin receptor-2 is overexpressed in the majority of malignant tumors, including neuroendocrine cancers, small cell lung cancer, breast cancer, and malignant lymphoma. The endogenous ligand of somatostatin receptors, somatostatin, has two isoforms of 14 and 28 amino acids, both of which demonstrate high binding affinity for somatostatin receptors. Owing to its short half-life in blood (2 min), imaging using somatostatin as a radiotracer is difficult. An analog of somatostatin, octreotide, which is formed from eight amino acid residues, has a longer half-life in blood (1.7 h) and higher metabolic stability than somatostatin. Many PET radiotracers derived from octreotide have been developed. Their chemical structures are illustrated in Fig. 1.6.

Fig. 1.6
PET radiotracers targeting somatostatin receptors: (a) octreotide, (b) [Tyr3]-octreotide, (c) [Tyr3,Thr8]-octreotide, (d) [1-NaI3]-octreotide, and (e) NOC-conjugated 1,4,7,10-tetraazacyclododecane
1.7.2 Integrin Receptor Subtype αvβ3
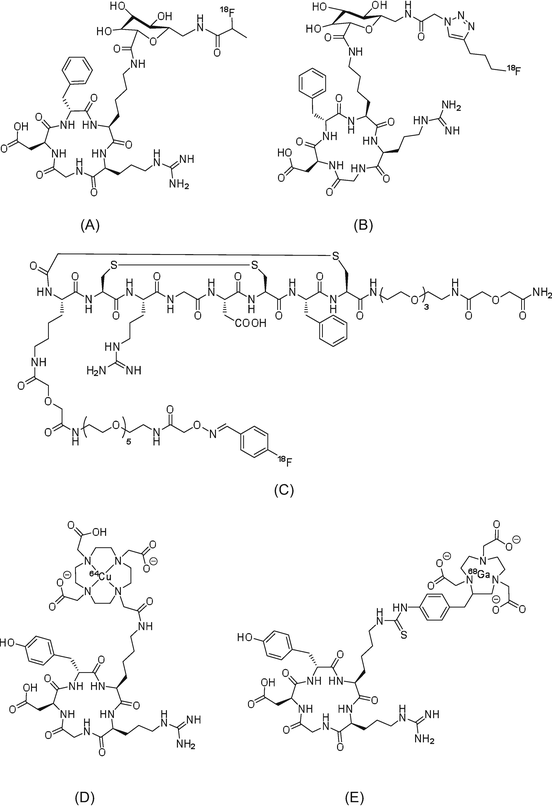
Angiogenesis is an important process during the proliferation of solid tumors. Cilengitide is a treatment developed to target integrin receptor (subtypes αvβ3 and αvβ5), which attenuates tumor angiogenesis. Almost all PET radiotracers targeting integrins contain the amino acid sequence, arginine-glycine-asparagine (RGD) [45, 46]. The RGD sequence, which is commonly found in extracellular matrix proteins, binds to integrins and shows particularly high affinity for the integrin αv subunit. Representative integrin-targeting PET radiotracers are 18F-, 68Ga-, and 64Cu-labeled compounds, including [18F]galacto-RGD, [18F]fluciclatide (AH111585), [18F]RGD-K5, 64Cu-DOTA-RGD, and 68Ga-NOTA-RGD (Fig. 1.7).
 Ethics, Regulations, and Clinical Development of Precision Medicine: Activating with Molecular Imaging
Ethics, Regulations, and Clinical Development of Precision Medicine: Activating with Molecular Imaging
 Clinical Evaluation of Focused High-Resolution Breast PET
Clinical Evaluation of Focused High-Resolution Breast PET
 Theranostic Approaches for Pathway-Activated Systems in Oncology
Theranostic Approaches for Pathway-Activated Systems in Oncology
 Recent Developments with Large-Bore PET/CT
Recent Developments with Large-Bore PET/CT
 Optical Imaging: How Far Can We Go
Optical Imaging: How Far Can We Go
 Applications of UIH High-Resolution PET/CT in Zhongshan Hospital
Applications of UIH High-Resolution PET/CT in Zhongshan Hospital




Related posts:
Ethics, Regulations, and Clinical Development of Precision Medicine: Activating with Molecular Imaging
Clinical Evaluation of Focused High-Resolution Breast PET
Theranostic Approaches for Pathway-Activated Systems in Oncology
Recent Developments with Large-Bore PET/CT
Optical Imaging: How Far Can We Go
Applications of UIH High-Resolution PET/CT in Zhongshan Hospital
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
