Fig. 43.1
Immunohistochemical staining for the estrogen receptor (ER) in breast cancer. The ER is detected as a brown color in the nucleus of the tumor cells (arrows).
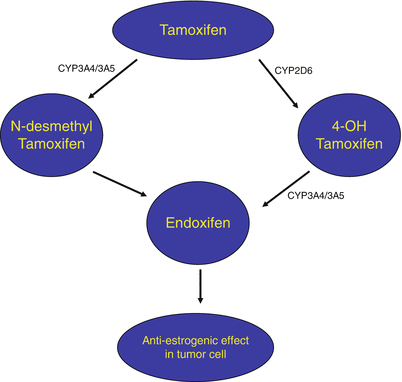
Fig. 43.2
Simplified schematic diagram showing metabolism of tamoxifen by CYP450 enzymes with the resulting production of the active metabolite, endoxifen.
43.4.2 UGT1A1
The uridine diphosphate glucuronosyl transferases (UGT) superfamily of endoplasmic reticulum-bound enzymes is responsible for conjugating a glucuronic acid moiety to a variety of compounds, thus allowing these compounds to be more easily eliminated. A member of this family catalyzes the glucuronidation of bilirubin, allowing it to be excreted in the bile. As irinotecan therapy for advanced colorectal cancers became more widely used, it was observed that patients who had Gilbert syndrome (defects in UGT leading to mild hyperbilirubinemia), suffered severe toxicity [30]. Irinotecan is converted to SN-38 by carboxylesterase-2, and SN-38 inhibits DNA topoisomerase I activity [31, 32]. SN-38 is glucuronidated by uridine diphosphate glucuronosyl transferase (UGT), forming a water-soluble metabolite, SN-38 glucuronide, which can then be eliminated (Fig. 43.3). The observation made in Gilbert syndrome patients revealed that SN-38 shares a glucuronidation pathway with bilirubin. The decreased glucuronidation of bilirubin and SN-38 can be attributed to polymorphisms in the UGT1A1 gene. The wild-type allele, UGT1A1*1, has six tandem TA repeats in the regulatory TATA box of the UGT1A1 promoter. The most common polymorphism associated with low activity of UGT1A1 is the *28 variant, which has seven TA repeats. Severe diarrhea and myelotoxicity can be found in those individuals who are homozygous for the *28 allele [33, 34]. In August 2005, the FDA amended the irinotecan (Camptosar®) package insert to recommend genotyping for the UGT1A1 polymorphism and suggested a dose reduction in patients homozygous for the *28 allele.
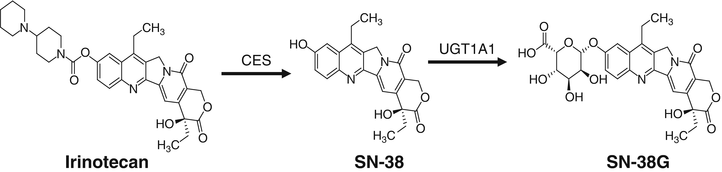
Fig. 43.3
Schematic diagram of irinotecan metabolism. The prodrug is converted to the active metabolite, SN-38, which is inactivated by UGT1A1.
43.4.3 TPMT
Thiopurine S-methyltransferase (TPMT) is a cytosolic enzyme that inactivates thiopurine drugs such as 6-mercaptopurine and azathioprine through methylation. Thiopurines are frequently used to treat childhood acute lymphoblastic leukemia and various other diseases including inflammatory bowel disease (IBD), and rheumatoid arthritis [35, 36]. They are also used as immunosuppressants in solid organ transplants. Thiopurines are metabolized partly by TPMT whose gene contains polymorphisms that result in null or decreased enzyme activity. These polymorphisms have been associated with increased risk of myelosuppression and toxicity.
Variability in activity levels of TPMT enzyme function exists between individuals, and it has been found that this variability can be attributed to polymorphisms of the TPMT gene [37, 38]. The most common variant alleles, TPMT*2, TPMT*3A, and TPMT*3C, account for 95 % of TPMT deficiency. Molecular technologies provide an accurate method for assessment of TPMT enzyme function in patients before treatment with thiopurines. Approximately 90 % of patients have the TPMT*1/*1 (wild-type) genotype and thus have normal TPMT enzyme activity. Up to 14 % of patients have been shown to be heterozygous for a TPMT genotype and possess one TPMT variant allele with lower enzyme activity. These patients may experience moderate to severe myelosuppression such that drug dose reduction may be warranted. Low TPMT activity levels could put a patient at risk for developing toxicity, since too much drug would be converted to 6-thioguanine nucleotides (6-TGNs), the cytotoxic active metabolite incorporated into DNA. On the other hand, a patient with high TPMT activity levels would need higher than standard doses of a thiopurine drug to respond well to the therapy, since a large amount of the drug is being inactivated before it can be converted to 6-TGNs. TPMT genotyping represents yet another example where molecular techniques can provide adequate information to assess drug-related risk of toxicity.
43.5 Pharmacogenomics and Drug Transporters
Although the genes that code for drug metabolizing enzymes have received more attention in recent years as markers for pharmacogenomic testing, the genes that code for proteins used to transport drugs across membranes also need to be considered when discussing a genetic basis of drug efficacy. These drug transporter proteins move substrates across cell membranes, bringing them into cells or removing them from cells. These proteins are essential in the absorption, distribution, and elimination of various endogenous and exogenous substances including pharmaceutical agents.
Several groups of drug transporters that may be significant in the field of pharmacogenomics exist, including multidrug resistance proteins (MDRs), multidrug resistance-related proteins (MRPs), organic anion transporters (OATs), organic anion transporting polypeptides (OATPs), organic cation transporters (OCTs), and peptide transporters (PepTs). ABCB1 (MDR1) is a member of the multidrug resistance protein family and is one example of a drug transporter protein important in the field of cancer pharmacogenomics.
43.5.1 ABCB1
ABCB1 is a member of the ATP-binding cassette (ABC) superfamily of proteins. Also known as P-glycoprotein (P-gp) or MDR1, it is a 170 kDa glycosylated membrane protein expressed in various locations including the liver, intestines, kidney, brain, and testis [39]. Generally speaking, ABCB1 is located on the membrane of cells in these locations and serves to eliminate metabolites and a wide range of hydrophobic foreign substances, including drugs, from cells by acting as an efflux transporter [40, 41]. Due to the localization of ABCB1 on specific cells, ABCB1 aids in eliminating drugs into the urine or bile and helps maintain the blood–brain barrier.
Like other eukaryotic ABC proteins, the ABCB1 protein is composed of two similar halves, each half containing a hydrophobic membrane-binding domain and a nucleotide-binding domain ([39]. The membrane-binding domains are each composed of six hydrophobic transmembrane helices and the two hydrophilic nucleotide-binding domains are located on the intracellular side of the membrane where they bind ATP. ABCB1 was first identified in cancer cells that had developed a resistance to several anticancer drugs because of an overexpression of the transporter. When expressed at normal levels in noncancerous cells, ABCB1 has been shown to transport other classes of drugs out of cells including cardiac drugs (digoxin), antibiotics, steroids, HIV protease inhibitors, and immunosuppressants (cyclosporin A).
Genetic variations in the ABCB1 gene expressed in normal cells have been shown to have a role in interindividual variability in drug response [39]. Although many polymorphisms have been detected in the ABCB1 gene, correlations between genotype and either protein expression or function have only been described for a few of the genetic variants. Most notable among these is the 3435 C > T polymorphism, found in exon 26, which has been found to result in decreased expression of ABCB1 in individuals homozygous for the T allele. The possible importance of the 3435 C > T is particularly interesting considering the mRNA levels are not affected by this polymorphism which is found within a coding exon. Although the correlation between the 3435 C > T polymorphism and ABCB1 protein levels may be due to linkage of this polymorphism to others in the ABCB1 gene, a recent study showed that this polymorphism alone does not affect ABCB1 mRNA or protein levels but does result in ABCB1 protein with an altered configuration. The altered configuration due to 3435 C > T is hypothesized to be caused by the usage of a rare codon which may affect proper folding or insertion of the protein into the membrane, affecting the function, but not the level of ABCB1 [42]. Genetic variations affecting the expression or function of drug transporter proteins, such as ABCB1, could drastically alter the pharmacokinetics and pharmacodynamics of a given drug.
43.6 Pharmacogenomics Applied to Oncology
Cancer represents a complex set of deregulated cellular processes that are often the result of underlying molecular mechanisms. While there is recognized inter-individual variability with respect to an observed chemotherapeutic response, it is also apparent that there is considerable cellular heterogeneity within a single tumor that may account for this lack of efficacy. It is becoming clear that molecular genetic variants significantly contribute to an individual’s response to a particular therapy and part of this response is due to polymorphisms in genes coding for drug-metabolizing enzymes as previously described. However, in the cancer patient the acquired genetics of the tumor cell must also be taken into account. Unlike traditional pharmacogenomic testing, testing for the cancer patient must be able to identify acquired or somatic genetic alterations that deviate from the underlying genome of the individual.
Cancer patients exhibit a heterogeneous response to chemotherapy with only 25–30 % efficacy. Pharmacogenomics can improve on chemotherapeutic and targeted therapy responses by providing a more informative evaluation of the underlying molecular determinants associated with tumor cell heterogeneity and by integrating information on drug responsiveness due to alterations in molecular biomarkers. The advancement of genotyping methods (i.e. next-generation sequencing) has allowed for the simultaneous identification of this heterogeneity. In large part these methods have illuminated genotyping as a major pharmacogenomics limitation. However, to date, only a handful of these mutations are deemed clinically actionable and have FDA-approved therapy associated with improved patients outcomes (Fig. 43.4) [20]. As research in clinical pharmacogenomics identifies more genotype specific therapies, the potential of these advanced genotyping methods in personalized medicine will be fully realized. Thus, the therapeutic management of the cancer patient can be tailored to include a specific therapy or a combination of therapies targeted to the patient’s tumor genotype.
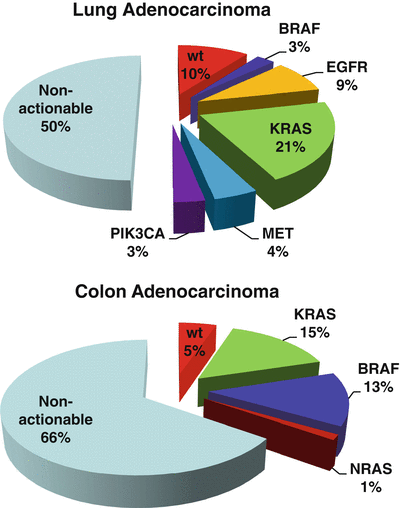
Fig. 43.4
Next-generation sequencing of cancers identifies actionable mutations. Types of mutations (actionable and non-actionable) identified in (a) lung adenocarcinomas and (b) colon adenocarcinomas by next-generation sequencing.
Most therapeutic drugs act on targets to elicit the desired effects. These targets include receptors, enzymes, or proteins involved in various cellular events such as signal transduction, cell replication, etc. Knowing if the target is present or being able to identify polymorphisms in these targets that either sensitize or render them resistant to the particular therapeutic agent is critical to patient management (Table 43.1). The following sections highlight several examples of characterized drug targets whose presence in a normal or variant form could affect the outcome of the therapeutic response.
Table 43.1
List of common mutation associated with the activation or resistance to FDA-approved pharmacogenomic therapies
Cancer type | Activity | Genes | Mutation | Therapeutic agent |
|---|---|---|---|---|
AML | Resistance | FLT3 | G835X | General TKl (sorafenib) |
Breast | Activating | HER2 | Amplification | HER2 antibodies (trastuzumab peruzumab) TKls (lapatinib) |
CML | Resistance | BCR-ABL1 | Y253H | ABL1 Tkl (lmatinib, nilotinib) |
E255K | ||||
E255V | ||||
V299L | ||||
F359C | ||||
F359l | ||||
F359V | ||||
T315A | ABL1 Tkl (imatinib, dasantinib) | |||
T315l | ||||
F317C | ||||
F317I | ||||
F317L | ||||
F317V | ||||
CRC | Resistance | KRAS | G12X | EGFR antibodies (cetuximab, panitumumab) |
G13X | ||||
Q61X | ||||
K117N | ||||
A146X | ||||
G12X | ||||
NRAS | Q61X | |||
BRAF | V600E | |||
Activating | BRAF | V600E | BRAF Tkls (vemurafenib, dabrafenib) | |
Melonma | Activating | BRAF | V600E | BRAF Tkls (vemurafenib, dabrafenib) |
NSCLC | Resistance | ALK-EML4 | L1196M | ALK/MET TKls (crizotinib) |
G1202R | ||||
S1206Y | ||||
1151Tins | ||||
L1152R | ||||
C1156Y | ||||
G1269A | ||||
EGFR | T700M | EGFR Tkls (erlotinib, gefitinib) | ||
Activating | EGFR | Exon 19 insertions | EGFR Tkls (erlotinib, gefitinib) EGFR/HER2 Tkls (afatinib) | |
Exon 19 deletions | ||||
Exon 20 insertions | ||||
L858R | ||||
G719X | ||||
L861Q | ||||
BRAF | V600E | BRAF Tkls (vemurafenib, dabrafenib) |
43.6.1 FLT3
FLT3 is a receptor tyrosine kinase expressed and activated in most cases of acute myeloid leukemia (AML) which has a relatively high relapse rate due to acquired resistance to traditional chemotherapies. An internal tandem duplication (ITD) mutation in the FLT3 gene is found in up to 30 % of AML patients while a point mutation at D835 has been shown to account for approximately 5 % of refractory AML [43, 44]. The FLT3-ITD induces activation of this receptor and results in downstream constitutive phosphorylation in STAT5, AKT, and ERK pathways. This mutation in FLT3 is a negative prognostic factor in AML.
FLT3-ITD-positive AML has become a clinical dilemma due to the poor results obtained with standard chemotherapy. One potential therapeutic option for patients who are at risk for relapse is allogeneic stem cell transplant [45]. In addition, randomized phase III trials using FLT3 inhibitors in combination with chemotherapy in patients with FLT3-positive AML are ongoing. Two promising multitargeted receptor tyrosine kinase inhibitors (TKIs), sorafenib and ABT-869, have been developed to suppress signal transduction from constitutively expressed kinases such as a mutated FLT3 [46]. The investigational FLT3-specific TKIs, quizartinib (AC220) and crenolanib, have also shown some recent success in a phase II trial in patients with FLT3-ITD+ AML with some evidence for increased activity against the D835 resistance mutation [47].
43.6.2 ALK
The fusion of the anaplastic lymphoma kinase (ALK) gene with the echinoderm microtubule-associated protein-like 4 (EML4) gene was first identified in 2007 in Japanese non-small-cell lung cancers (NSCLCs) [48]. The EML4-ALK fusion gene is the result of an inversion on the short arm of chromosome 2 that joins exons 1–13 of EML4 to exons 20–29 of ALK [49]. The fusion gene can be detected by use of break-apart probes and FISH technology (Fig. 43.5).
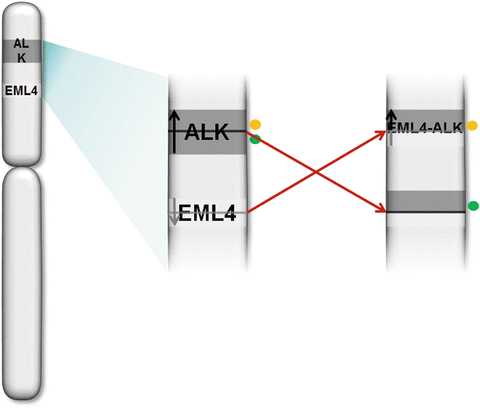
Fig. 43.5
EML4- ALK FISH .Diagram of translocation and location of break-apart probes (colored dots).
The EML4-ALK fusion gene represents a novel biomarker and potential molecular target in NSCLC and translocations or rearrangements of the ALK gene with other genes have been reported in several other human cancers [49, 50]. Approximately 3–5 % of NSCLCs found in North America harbor the EML4-ALK fusions. The fusion of these two genes results in constitutive ALK tyrosine kinase activity which has significant oncogenic function. Because of its oncogenic role, the EML4-ALK fusion product has been a target for tyrosine kinase inhibitors. Several ALK kinase inhibitors have been developed and FDA-approved for treatment of NSCLC, such as crizotinib (Xalkori) and certitnib (Zykadia) [49, 51]. Crizotinib is a first-line oral inhibitor of ALK tyrosine kinase activity which has shown high response rates in patients with EML4-ALK-positive NSCLC [52]. However, a large number of patients treated with crizotinib develop resistance to treatment, many by acquiring secondary mutations in the ALK tyrosine kinase domain, driving relapse [53]. These patients are now eligible for treatment with a newly approved ALK TKI, ceritinib, which has exhibited a response rate of greater than 70 % in those patients resistant to crizotinib [54]. In addition, there are numerous phase II trials investigating the efficacy of heat shock protein 90 (HSP-90) inhibitors to treat patient who are positive for ALK fusions. Early results suggest a significant response to treatment with these HSP-90-targeted inhibitors [55]. The identification of these resistance mutations and the successful development of second- and third-generation therapies to combat them exhibit the utility in fully characterizing the genotypes of therapy-resistant tumors.
Related posts:




Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


