Chapter 66 Pediatric Soft Tissue Sarcomas
Epidemiology
The soft tissue sarcomas account for approximately 7% of all pediatric cancers. Rhabdomyosarcoma (RMS) comprises 40% of this group and the other nonrhabdomyosarcoma soft tissue sarcomas (NR-STS) comprise the remainder.1,2 This results in 350 patients with RMS and 500 with NR-STS (divided about 2 : 1 between high and low grade) available for participation in clinical trials in the United States annually. Based on prior clinical trials, 75% of children with RMS have required radiation therapy (RT), and, based on the presence of high-grade disease only, approximately two thirds of children with NR-STS would receive radiation. This yields 600 sarcoma cases annually for the study of radiation-specific clinical trial questions, training of young radiation oncologists, and study of radiation-specific long-term sequelae.
Patients with RMS have benefited from the Children’s Oncology Group (COG) sarcoma studies (formerly the Intergroup Rhabdomyosarcoma Study Group [IRSG]). The trials conducted by these groups have defined the combined modality management of children with RMS in North America and allowed the randomized comparisons of new chemotherapeutics as well as RT strategies that would not be possible without the cooperative group structure. These approaches, as well as the specifics of modern RT are discussed in this chapter. Treatment of children with NR-STS has been less well defined. Local therapy approaches have evolved from adult paradigms, incorporating limb salvage over amputation, adjuvant irradiation, and, subsequently, preoperative RT approaches.3–6 Despite an adult parallel to draw from, little prospective research has been conducted in the pediatric NR-STS population. An ongoing trial through COG (ARST0332) seeks to address some of these deficiencies delivering both preoperative and postoperative RT in conjunction with surgery and chemotherapy to define the role of these local and systemic approaches in a comprehensive clinical trial.
Biologic Characteristics and Pathology
Rhabdomyosarcoma
RMS is broadly classified as one of the small, round, blue cell malignancies of childhood.7 The histologic subtypes, in order by worsening prognosis, include embryonal rhabdomyosarcoma (ERMS), the most common form (with spindle cell and botryoid comprising two favorable variants), alveolar rhabdomyosarcoma (ARMS), and undifferentiated sarcoma. Patients with undifferentiated sarcoma were previously enrolled on the IRSG clinical trials in a fashion similar to ARMS. Currently, these patients receive systemic therapy similar to patients with Ewing’s sarcoma. Light microscopy often describes ERMS with spindle-shaped cells and ARMS with small, round, blue cells forming alveolar-like spaces, although tumor biopsy specimens may also look like collections of poorly differentiated cells complicating definitive diagnosis by hematoxylin and eosin staining alone. Immunohistochemical staining can include positivity for MyoD.8 Cytogenetic events occur in ERMS with a loss of heterozygosity on chromosome 11p and in ARMS with translocations between chromosomes 2 and 13 or 1 and 13. These represent loci of PAX3 and PAX7 (chromosomes 2 and 1) and their fusion with FKHR on chromosome 13. These fusion proteins act as transcription factors and are diagnostic of ARMS.7 Other genetic abnormalities and syndromes associated with RMS include neurofibromatosis type 1 (with a prevalence of 1 : 200 noted on IRSG-IV), Li-Fraumeni syndrome, and Beckwith-Wiedemann syndrome.9–11
Nonrhabdomyosarcoma Soft Tissue Sarcoma
The pediatric NR-STS comprise a heterogeneous group of tumors managed homogeneously owing primarily to the limitations in both patient numbers and effective chemotherapy. The most common histologies noted in practice and clinical trials include synovial cell sarcoma and malignant peripheral nerve sheath tumor (MPNST), although several rarer variants including rhabdoid tumor and infantile fibrosarcoma are almost exclusive to the pediatric age group.12,13 Although many NR-STSs have no genetic signature, several histologic variants do have specific translocations or deletions12,14 (Table 66-1).
TABLE 66-1 Genetic Abnormalities: Nonrhabdomyosarcoma Soft Tissue Sarcomas
| Histology | Genetic Abnormality |
|---|---|
| Synovial cell sarcoma | t(X;18); fusion SYT-SSX |
| Clear cell sarcoma | t(12;22); fusion EWS-ATF1 |
| Rhabdoid tumor | Deletion 22q; INI1 deletion |
| Desmoplastic small round cell tumor | t(11;22); fusion EWS-WT1 |
| Infantile fibrosarcoma | t(12;15); fusion ETV6-NTRK3 |
| Alveolar soft part sarcoma | der(17)t(X;17); fusion TFE3-ASPL |
| Low-grade fibromyxoid sarcoma | t(7;16); fusion FUS-CREB3L2 |
Clinical Manifestations
Rhabdomyosarcoma
The presentation of children with RMS is diverse and relates to the site of involvement and extent of disease. The median age for children presenting with RMS is younger than 5, although another peak occurs in the mid teens. The head and neck region is the most frequent site of involvement and is divided into favorable and unfavorable (parameningeal) sites.15,16 This site comprised 42% of localized presentations in patients enrolled on IRSG III and IRSG IV and serves as an example of the important nature of primary tumor location.17 Metastatic disease occurs 20% of the time at presentation, and nodal involvement is rare (<5%) except for paratesticular (25%) and extremity (24%) sites of disease, which warrant either computed tomography (CT) (paratesticular, <10 years of age), nodal sampling (paratesticular, ≥10 years of age), or sentinel node biopsy (extremity site).18–21
Nonrhabdomyosarcoma Soft Tissue Sarcoma
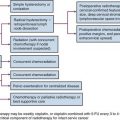
The presentation of NR-STS is often as a painless mass whereas other symptoms are site specific. Approximately half of the cases arise in the extremity, and the incidence increases throughout the adolescent years and into adulthood.22,23 Although the vast majority of soft tissue “masses” are either not real or benign, consideration should be given to malignancy because an ill-chosen surgical approach may compromise future local therapy (Fig. 66-1).
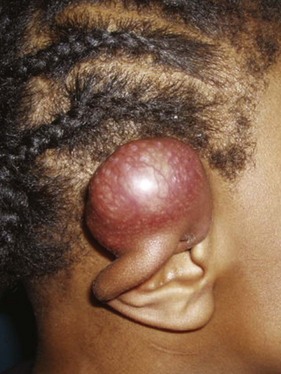
Fifteen to 22 percent of patients present with metastatic disease at diagnosis, with the lungs being the predominant site of metastatic involvement.13,24 Clear cell sarcoma, epithelioid sarcoma, and angiosarcoma carry a risk of regional nodal involvement and warrant evaluation of the draining nodal bed(s) with sentinel node sampling.
Staging
Rhabdomyosarcoma
Staging workup is similar for most patients with RMS (Table 66-2). A site-specific history and physical examination should be performed by the treating radiation oncologist at the time of presentation to define the site of primary disease, its extent, and symptoms related to potential metastatic sites of involvement. Attention to cranial nerve involvement for head and neck primary sites as well as attending the examination under anesthesia for genitourinary sites (vaginal, cervix, uterus, bladder, and prostate) will assist in the multidisciplinary discussion regarding staging and local therapy approaches.
TABLE 66-2 Staging Workup and Follow-Up Evaluations for Patients with Rhabdomyosarcoma and Nonrhabdomyosarcoma Soft Tissue Sarcomas
| Rhabdomyosarcoma | Nonrhabdomyosarcoma Soft Tissue Sarcomas | |
|---|---|---|
| Staging | ||
| Clinical | Site-directed history and physical examination | Site-directed history and physical examination |
| Imaging | MRI of primary tumor | MRI of primary tumor |
| CT of chest/abdomen | CT of chest | |
| Bone scintiscan | ||
| Procedures | Biopsy: primary tumor/metastasis | Biopsy: primary tumor/metastasis |
| Extremity: sentinel lymph node biopsy | Histology: specific sentinel lymph node biopsy | |
| Head and neck: cerebrospinal fluid cytology | ||
| Genitourinary: examination under anesthesia | ||
| Follow-up | ||
| Year 1 | History and physical examination every 3 months, laboratory studies, MRI of primary tumor area | History and physical examination, laboratory studies every 3 months, MRI of primary tumor area |
| CT of chest and other metastatic imaging every 3 months | CT of chest every 6 months | |
| Years 2 to 3 | Every 4 months: history and physical examination, laboratory studies, MRI of primary area | Every 5 months: history and physical, laboratory studies, MRI of primary tumor region, CT of chest |
| Every 4 months: CT of chest and other metastatic imaging | ||
| Years 4 to 5 | Every 6 months: history and physical, MRI of primary area, chest radiograph | Annual history and physical laboratory studies, MRI of primary area, CT of chest |
After staging evaluation, a clinical stage should be assigned (outlined in Table 66-3). If surgery has been performed (or after a planned surgical resection or biopsy), a grouping should be defined (Table 66-4). These two factors plus the patient’s histology are combined into a risk classification to assign patients to protocol therapy.
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


