Pathophysiology of Atherothrombosis
Peter Libby
Paul M. Ridker
Kevin Croce
Advances in contemporary vascular biology have contributed enormously to increased understanding of the pathogenesis of atherosclerosis and its thrombotic complications, and have increased our understanding of the mechanisms by which therapeutic interventions can alter the course of this disease. This chapter reviews some of this progress, highlighting the emerging links between atherogenesis, inflammation, and thrombosis: three processes classically considered separately. Indeed, the tendency to view atherosclerosis and thrombosis as distinct has deep historical roots. During the mid-19th century, Karl von Rokitansky1 postulated that incorporation of thrombi into the artery wall caused atherosclerosis; the celebrated cellular pathologist Rudolph Virchow2 ridiculed this view. Virchow considered atherosclerosis a disease predicated on proliferation and death of vascular wall cells, complicated by cholesterol accumulation and inflammation. Virchow’s eminence deterred acceptance of the relation between thrombosis and atherogenesis.
The role of thrombosis in atherogenesis has resurfaced periodically since the time of these pioneers. Duguid,3 in the middle of the 20th century, revived von Rokitansky’s view that accorded a key role to the incorporation of thrombi in the formation of atheromatous lesions. Hand and Chandler4 found that induction of thrombi could induce lesions resembling atheroma in experimental animals. Various investigators who used increasingly sophisticated biochemical methods, including Smith and Bini, localized products of thrombosis such as fibrin in human atheromatous plaques.5,6
We now recognize that both views in this polarized debate apply to the pathobiology of atherosclerosis, and that thrombotic and inflammatory cellular pathways are inextricably intertwined. Although the very earliest lesions of atherosclerosis may not involve platelets or coagulation, thrombosis nonetheless contributes to all subsequent stages of atherosclerosis. Indeed, some experimental evidence suggests a role for platelets even in early murine atherosclerosis.7 Moreover, compelling laboratory and clinical evidence now provides growing support for the role of inflammation in both atherogenesis and the pathophysiology of its thrombotic complications. This discussion highlights the interactions between inflammation, thrombosis, and atherosclerosis during various stages of atherogenesis.
THREE THEMES IN ATHEROTHROMBOSIS
Several themes recur while considering the interfaces between atherogenesis, inflammation, and thrombosis. First, the pathogenic effects of thrombosis in the context of atherogenesis reflect dysregulation of fundamental biologic mechanisms of importance in normal homeostasis. The products of coagulation can alter vascular wall cells function in a manner that promotes fibrosis and healing. Substances released from activated platelets, such as platelet-derived growth factor (PDGF) and transforming growth factor-β (TGF-β), can augment the synthesis of collagen by smooth muscle cells (SMCs).8 PDGF is a potent chemoattractant that stimulates the migration of SMCs.9,10,11,12 Thrombin, generated during the clotting cascade, is both a smooth muscle mitogen and a proinflammatory mediator that regulates the expression of many other genes that influence vascular injury responses.13,14,15,16 All of these processes may prove essential during wound healing and tissue repair. Yet, these usually salutary functions can become pathogenic in the context of the artery wall. The interface of atherosclerosis with thrombosis invokes the same conundrum that applies to the inflammatory aspects of atherosclerosis. Normal homeostatic mechanisms expressed inappropriately or in excess actually contribute to the pathogenesis of plaques.
A second recurring theme concerns the balance between stimulatory and inhibitory limbs of regulatory pathways. This concept, pertaining to the opposing poles of proteases versus antiproteases, or thrombosis versus fibrinolysis, is quite familiar to students of thrombosis. We shall see subsequently how altered balances between such pathways contribute to atherogenesis.
A third recurring theme has emerged more recently. We increasingly recognize that inflammation can heighten thrombosis and that mediators of thrombosis can play roles not only in clot formation, but also in augmenting inflammation (FIGURE 89.1). For example, thrombin can elicit inflammatory mediators, notably CD40 ligand (CD154), which can induce tissue factor expression on vascular cells and macrophages (MF), the principal source of this potent procoagulant in the atheroma.17,18 In turn, platelets, when activated, can release CD40 ligand; myeloid-related protein 14 (MRP-14), regulated on activation, normal T cell expressed and secreted (RANTES); and other proinflammatory mediators.19,20 Inflammatory mediators can sensitize vascular muscle and endothelial cells (ECs), as well as mononuclear phagocytes, to undergo programmed cell death or apoptosis. Dying vascular cells and macrophages can elaborate tissue factor-rich microparticles that can promote thrombosis. Therefore, reciprocal reinforcement of inflammation by thrombosis, and vice versa, results in amplification of these pathologic processes.
RISK FACTORS FOR ATHEROSCLEROSIS
Work conducted over the last half century has defined several risk factors for development of atherosclerosis and its complicating events. The concept of risk factors has gained widespread
clinical application in generating algorithms for predicting an individual’s likelihood of having a future cardiovascular event. The concept of risk factors has led to interventions to reduce cardiovascular events. In the context of the present consideration of pathophysiology, the relationship of certain factors of risk to atherosclerosis and its clinical manifestations suggests hypotheses regarding mechanisms of development of the disease and the mechanisms of its complicating events. A complete exposition of risk factors, risk prediction algorithms, and intervention studies exceeds the scope of this chapter. Many authoritative reviews treat these subjects.21 This overview of the risk factors for atherosclerosis will consider traditional risk factors, both nonmodifiable and modifiable, as well as emerging risk factors.
clinical application in generating algorithms for predicting an individual’s likelihood of having a future cardiovascular event. The concept of risk factors has led to interventions to reduce cardiovascular events. In the context of the present consideration of pathophysiology, the relationship of certain factors of risk to atherosclerosis and its clinical manifestations suggests hypotheses regarding mechanisms of development of the disease and the mechanisms of its complicating events. A complete exposition of risk factors, risk prediction algorithms, and intervention studies exceeds the scope of this chapter. Many authoritative reviews treat these subjects.21 This overview of the risk factors for atherosclerosis will consider traditional risk factors, both nonmodifiable and modifiable, as well as emerging risk factors.
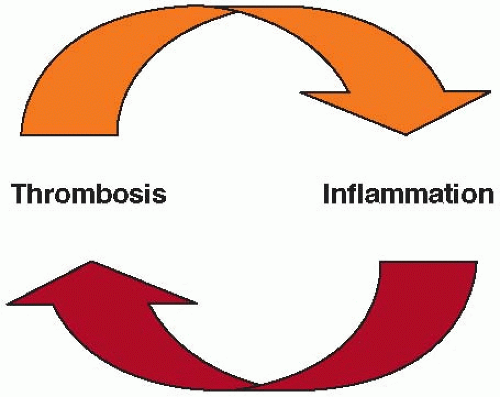 FIGURE 89.1 Accumulating data linking inflammation and thrombosis support the scheme illustrated here, that inflammation begets thrombosis and vice versa. An untested corollary of this concept posits that anti-inflammatory therapies may limit thrombosis, and that antithrombotic therapies may reduce vascular inflammation. (From Libby P, Simon DI. Inflammation and thrombosis: the clot thickens. Circulation 2001;103:1718-1720, with permission.) |
Nonmodifiable Risk Factors for Atherosclerosis
Age
Age predicts most potently an individual’s risk for an atherosclerotic event.22 This recognition has important implications, given the anticipated accumulation of the proportion of elderly individuals in many developed countries. From the perspective of thrombosis, the increase in older individuals with atherosclerosis has important implications, as these individuals have higher risk of adverse effects from some therapeutic interventions, including antiplatelet and anticoagulant therapy.
Sex
Sex strongly influences cardiovascular risk. Although women enjoy relative protection from cardiovascular risk premenopausally, their cardiovascular risk increases following menopause. The accumulation of elderly individuals in many populations alluded to above will yield a preponderance of elderly women as an important pool of cardiovascular risk in coming decades. Despite a considerable body of observational data that supported a protective role of hormone therapy against atherosclerotic disease in women, large-scale randomized clinical trials have not affirmed reductions in cardiovascular event rates in women receiving hormonal therapy, particularly in those with established disease.23 Indeed, several of these studies have shown elevated risk of thrombotic events in women receiving estrogen or estrogen plus progesterone supplementation.23
Family History and Genetic Predisposition
A family history of coronary heart disease before age 60 in a parent adds to the other traditional risk factors in estimating the likelihood of a cardiovascular event.24,25. Some single-gene mutations clearly contribute to enhanced cardiovascular risk—for example, those that cause loss of function of the low-density lipoprotein (LDL) receptor, such as familial hypercholesterolemia, familial defective apo B, or autosomal dominant hyperlipoproteinemia. Yet these well-understood monogenic causes of atherosclerosis contribute a small portion of population-attributable risk of this disease. Thus, genetic predisposition to atherothrombotic events appears to depend on polygenic inheritance, with each variant contributing a small increment in risk. Most single-nucleotide polymorphisms associated with coronary artery disease have proved poorly reproducible or generalizable.26
The introduction of genome-wide association approaches has, in contrast, led to reproducible identification of regions that confer cardiovascular risk in human populations. Notably, the chromosome 9p21 locus consistently associates with augmented cardiovascular event rates in several populations.27,28,29 The exploitation of the results of genome-wide association scans for atherosclerosis (and many other chronic diseases) should yield exciting new insights into pathophysiology and potential therapeutic targets in the future. From a practical standpoint at present, however, a simple clinical query regarding family history of premature coronary artery disease appears to confer as much information regarding risk as an assortment of newly emerged genetic markers.30 Thus from a clinical perspective, genetic testing today adds little to cardiovascular risk assessment over and above the traditional risk factors, including a simple query regarding family history. The 2010 American Heart Association (AHA) guidelines for risk assessment in asymptomatic adults reflect this conclusion.31
Modifiable Risk Factors for Atherosclerosis
Lipids
Of the modifiable risk factors, lipids have led the way. The appreciation of links between cardiovascular risk, atherosclerosis, and hypercholesterolemia emerged over the last century. In particular, LDL, a major carrier of cholesterol in humans, participates causally in atherogenesis (FIGURE 89.2). LDL levels correlate with cardiovascular risk in populations.32 Genetic abnormalities that yield excess LDL correlate with increased individual risk of cardiovascular events.33 Interventions that lower LDL confer protection from cardiovascular events.34 A large body of experimental data supports a causal role for LDL in altered cellular events that sow the seeds for atherosclerosis.35,36 For example, excess LDL correlates with accumulation in the arteries of lipid-laden macrophages, cells that participate importantly in atherothrombosis. Moreover, lowering cholesterol levels by diet alone or by statin treatment can decrease expression of tissue factor within plaques, the procoagulant implicated in triggering many atherothrombotic events.37
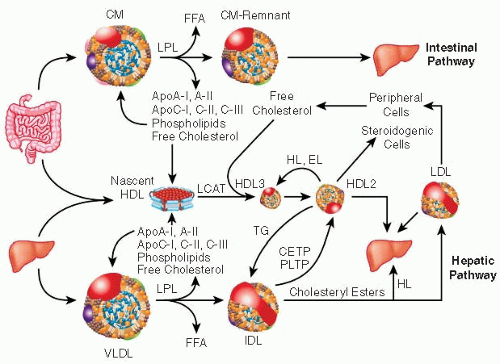 FIGURE 89.2 Schematic diagram of the lipid transport system. EL, endothelial lipase; HL, hepatic lipase. (From Genest J, Libby P. Lipoprotein disorders and cardiovascular disease. In: Bonow RO, Mann DL, Zipes DP, et al., eds. Braunwald’s heart disease: a textbook of cardiovascular medicine, 9th ed. Philadelphia, PA: Elsevier Saunders, 2011:975-995, copyright Elsevier, with permission.) |
Low-Density Lipoprotein
LDL particles exhibit heterogeneity. Small, dense LDL particles confer particular risk and associate with high levels of triglycerides and low levels of high-density lipoprotein (HDL), a triad characteristic of “diabetic dyslipidemia.” Larger and less dense LDL particles appear less likely to provoke clinical complications. Several advanced tests can characterize lipoprotein particles. In particular, measuring the levels of LDL’s major apolipoprotein, apolipoprotein B (apo B), provides an excellent estimate of particle number, as each LDL bears one apo B moiety. Canadian guidelines recognize apo B measurements as appropriate for clinical use. Yet, for most practitioners, inspection of the lipid profile can provide sufficient information for the management of most patients, without the need for advanced testing.31
Inhibitors of the rate-limiting enzyme in cholesterol synthesis, hydroxymethylglutaryl coenzyme A—a class of drugs known as statins—strikingly lower LDL levels. These drugs likely accomplish LDL lowering largely by increasing levels of LDL receptors and enhancing clearance of LDL. Treatment with statins has lowered cardiovascular event rates in broad categories of patients, ranging from those who have already sustained myocardial infarctions (MIs) with high levels of LDL to individuals with no prior history of cardiovascular disease and average or even below-average levels of LDL.34 Statins undoubtedly reduce cardiovascular events through LDL lowering, but this class of drugs also alters prenylation of small G proteins such as ras, rac, or rho, important in controlling functions of cells involved in atherosclerosis.38 Statinmediated reduction of rho function limits the production of prothrombotic microparticles from ECs.39 Statins also activate the transcription factor Kruppel-like factor 2 (KLF2), which regulates a number of inflammatory functions of ECs, including thrombomodulin gene expression.40,41 Thus statins may also produce cardiovascular benefit through pathways independent of LDL lowering. Subsequent sections will deal at greater length with putative anti-inflammatory actions of statins.
Triglycerides
Triglycerides have engendered considerable controversy as independent risk factors for atherosclerosis and its complications. Triglyceride-rich lipoproteins include very low-density lipoprotein, a lipoprotein that like LDL, bears apo B as its signature apolipoprotein. Levels of triglycerides typically rise following meals, particularly those rich in fat. Recent studies, however, have indicated that random measurements of triglyceride levels serve well as estimates of future cardiovascular risk, obviating the need for obtaining fasting levels in clinical practice.42
Although triglyceride levels correlate well with prospective cardiovascular risk, when adjusted for covariates such as levels of HDL, the incremental risk conferred by triglycerides appears negligible.43 Moreover, clinical trials have not shown that interventions that lower triglyceride levels, such as fibric acid derivatives, consistently lower cardiovascular events or mortality even in diabetic individuals who have a predisposition to hypertriglyceridemia.44,45
High-Density Lipoprotein
Abundant observational data have established low HDL as a consistent and important risk factor for atherosclerosis. From a pathophysiologic perspective, HDL may mitigate atherogenesis through severalmechanisms. As proposed originally by Glomset,46 HDL particles can mediate reverse cholesterol transport, ferrying cholesterol out of cells such as macrophages. Experimental data support the ability of HDL particles to efflux cholesterol from lipid-loaded macrophages. Apolipoprotein A-I (apo A-I) interacts with the ATP-binding cassette transporter ABCA1, a process important in generating nascent HDL particles. Mature HDL particles can efflux cholesterol using a “half” transporter, homologous to ABCA1, known as ABCG1.47 Animal experiments have documented HDL’s function in cholesterol efflux from storage sites to bile acids and ultimately the feces,48 but only a small fraction of total body reverse cholesterol transport probably occurs from atheromatous lesions in humans. Moreover, HDL efflux from macrophages appears to be independent of HDL itself as a determinant of vascular disease and thus may provide a link between HDL, thrombosis, and inflammation.49
In addition to functioning as an acceptor for reverse cholesterol transport, HDL carries many proteins that may help modulate aspects of atherothrombosis, locally in plaques and in innate immunity systemically.50 In addition to a long appreciated binding capacity for bacterial endotoxin, recent proteomic analyses have demonstrated that HDL associates with modulators of proteinase activity, complement activation, lipid metabolism, and inflammation.51 Experimental studies both in vitro and in vivo determined that HDL may exert anti-inflammatory actions relevant to atherothrombosis.52 Despite this biological plausibility, a large Mendelian randomization study has not supported HDL as a causal risk factor in atherosclerosis.53
Although HDL levels indubitably correlate with risk for atherosclerotic events, interventions that raise HDL have not yet proven effective in reducing cardiovascular risk.54 Clinical trials are evaluating various strategies for raising HDL in patients at risk for cardiovascular disease. Inhibition of cholesteryl ester transfer protein (CETP) raises HDL levels. Yet, one inhibitor of CETP provoked harm rather than benefit in a large clinical trial.55 The particular agent used in this study, torcetrapib, exerted mineralocorticoid agonist properties that raised blood pressure, lowered potassium, and may have contributed to adverse outcomes. A large clinical endpoint study with another CETP inhibitor, dalcetrapib, halted in 2012 apparently for futility. Two CETP inhibitors that appear not to share the off-target actions of torcetrapib (anacetrapib and evacetrapib) are currently in clinical development. The results of trials with these agents should establish whether CETP inhibition to raise HDL could yield clinical benefit.56
Fibric acid derivatives (agents such as fenofibrate, gemfibrozil, or bezafibrate) weakly induce apo A-I transcription through activation of peroxisomal proliferation activating receptor α.57 This family of drugs can substantially lower triglycerides and slightly raise HDL. A well-understood drug-drug interaction precludes the safe combination of gemfibrozil with statins, agents mandated for use in most individuals eligible for pharmacotherapy of lipid disorders.58 Several recent studies with fenofibrate have failed to show benefit on cardiovascular outcomes or mortality in people with diabetes, as noted above.44,45 The clinical role of fibrates in treating individuals with low HDL levels, particularly those already receiving statins, remains unproven.
Nicotinic acid substantially raises HDL levels, and when combined with statins, produces highly favorable, effects on the lipid profile, raising HDL and lowering LDL. While some older clinical trial data show reduction in MI in individuals treated with nicotinic acid, the added benefit of nicotinic acid to the contemporary standard of care remains uncertain. One recent trial (AIM-HIGH) stopped early due to futility [NEW REFERENCE T/K]. Another clinical trial currently in progress is testing whether combinations of statins with sustained release or nicotinic acid and an inhibitor of prostaglandin D intended to reduce flushing may provide added benefit over the current standard of care [NEW REFERENCE T/K].
Hypertension
In addition to lipids, high blood pressure constitutes a major modifiable risk factor. Actuarial data established striking relationships between mortality and blood pressure levels at a time when many physicians thought that hypertension represented an adaptive response to arteriosclerosis that inevitably accompanied aging. Indeed, until the last few decades, many considered hypertension abeneficial adaptation to aging arteries. Consistent cohort studies and cross-sectional data have established the importance of even slight elevations in blood pressure as a contributor to cardiovascular risk.59 Current thinking accords a primary role to systolic blood pressure as a determinant of cardiovascular risk, although pulse pressure also independently associates with elevated cardiovascular risk. Some elegant analyses of relatively rare genetic abnormalities have inculcated abnormalities in renal electrolyte handling as etiologic in hypertension.60 Yet the vast majority of patients with hypertension do not have monogenic or well-defined endocrine causes of elevated blood pressure.
Intervention studies have established blood pressure lowering as an important public health goal and an avenue to reducing individual cardiovascular risk.61 In particular, pharmacologic intervention to reduce blood pressure lowers the risk of cerebrovascular accidents. Although more difficult to establish, current data indicate reduced MI as a benefit of blood pressure control. While dietary measures (as illustrated by the DASH diet) and other lifestyle interventions (such as engaging in regular physical activity) can help control blood pressure,62 most individuals with established hypertension will also require pharmacotherapy. Several large-scale clinical trials have compared various antihypertensive drugs, and this ensemble of data ultimately suggests that for the most part, effective blood-pressure lowering— regardless of the particular agents used—can lower cardiovascular events. Distilling recent clinical trials for some practical precepts, a thiazide-type diuretic should be part of most antihypertensive regimens. Current practice employs lower doses of thiazides than used previously—12.5 or 25 mg of chlorthalidone or hydrochlorothiazide daily, for example.63 Beta-adrenergic blocking agents should be used primarily in individuals who have already sustained an MI, and generally should not be firstline antihypertensive agents for others. Black individuals may benefit more from calcium channel blockers than from angiotensin-converting enzyme (ACE) inhibitors. Clinical trials have established no clear-cut benefit of angiotensin receptor blockers (ARBs) over ACE inhibitors in preventing atherothrombotic events, and there is ongoing controversy on whether ARB use associates with increased risk of maliganancy.64 Studies do not support the combination of ACE inhibitors and ARBs.
Cigarette Smoking
Active cigarette smoking confers considerable cardiovascular risk.65 Mechanisms by which cigarette smoking augments atherothrombotic events remain uncertain, although observational and experimental data provide some hints in this regard. Cigarette smoking may augment oxidative stress, thought to promote atherogenesis. Cigarette smoking can also lower HDL. Smoking cessation leads to a rapid decline in cardiovascular event rates, yielding benefits in <1 year. As many consider this too short a period to modify atherosclerotic plaques per se, some have interpreted this rapid return toward baseline event rates after smoking cessation as an indication that smoking particularly heightens thrombotic diathesis. While the mechanisms remain unproven, smoking cessation should be a priority of practice and public health intervention.
Emerging Risk Factors for Atherosclerosis
In addition to the traditional risk factors enumerated above, several more recently recognized risk markers have come to the fore as potential tools for sharpening risk prediction, and
perhaps shedding mechanistic light on the pathogenesis of atherothrombosis. One such emerging risk factor, hyperhomocysteinemia, has particular relevance to thrombosis.66 Monogenic defects that lead to hyperhomocysteinemia, such as defects in beta-cystathionine synthase, associate with a thrombotic diathesis in childhood. More modest elevations of homocysteine in adult populations also associate with elevated atherothrombotic risk, although when adjusted for confounders, high levels of homocysteine account for only a modest enrichment in the relative risk of cardiovascular disease.67,68 Folic acid administration can lower homocysteine levels. Recent trials have evaluated the efficacy of folate supplementation in reducing cardiovascular events. These studies have uniformly shown no benefit from such vitamin therapy, despite lowering of homocysteine.69,70,71 As grain and cereal products currently undergo folate enrichment in the United States, as part of a public health intervention to reduce neural tube defects, pharmacologic intervention to lower cardiovascular risk by lowering homocysteine levels with folate or other vitamins appears unwarranted.
perhaps shedding mechanistic light on the pathogenesis of atherothrombosis. One such emerging risk factor, hyperhomocysteinemia, has particular relevance to thrombosis.66 Monogenic defects that lead to hyperhomocysteinemia, such as defects in beta-cystathionine synthase, associate with a thrombotic diathesis in childhood. More modest elevations of homocysteine in adult populations also associate with elevated atherothrombotic risk, although when adjusted for confounders, high levels of homocysteine account for only a modest enrichment in the relative risk of cardiovascular disease.67,68 Folic acid administration can lower homocysteine levels. Recent trials have evaluated the efficacy of folate supplementation in reducing cardiovascular events. These studies have uniformly shown no benefit from such vitamin therapy, despite lowering of homocysteine.69,70,71 As grain and cereal products currently undergo folate enrichment in the United States, as part of a public health intervention to reduce neural tube defects, pharmacologic intervention to lower cardiovascular risk by lowering homocysteine levels with folate or other vitamins appears unwarranted.
Lipoprotein a [Lp(a)]
Lp(a), pronounced “L P little A,” denotes a subclass of LDL characterized by an apo B moiety covalently bound to apolipoprotein a. Curiously, Apo(a) bears striking homology to plasminogen, including a variable number of repeats of the kringle structure characteristic of plasminogen, particularly kringle 4. Despite the sequence similarity to plasminogen, Apo(a) appears to lack enzymatic capacity. The distribution of Lp(a) levels in the population is skewed. Those with high levels of Lp(a) have heightened risk of atherothrombotic events. Genome-wide association studies and Mendelian randomization studies support a causal role for Lp(a) in atherothrombosis.72 Over an entire population, due to the skewed distribution of Lp(a) levels, Lp(a) accounts for a small portion of events (a low population-attributable risk). In individuals with higher levels of Lp(a), however, the incremental risk may substantially elevate the risk of atherothrombotic events. Unfortunately, specific interventions to lower Lp(a) are few, notably nicotinic acid treatment. Studies designed to lower Lp(a) have not been conducted. Current management strategies for those with high levels of Lp(a) include LDL lowering by lifestyle measures and statin therapy.
Oxidative Stress
A great deal of preclinical data and biomarker studies in human populations indicate a role for oxidized low-density lipoprotein in the pathogenesis of atherothrombosis.73 Yet, a variety of interventions aimed at reducing LDL oxidation have failed to produce clinical benefit. Several studies with antioxidant vitamins have shown no improvement in cardiovascular event rates. A lipophilic antioxidant agent that effectively reduced LDL oxidation experimentally, likewise failed to reduce cardiovascular endpoints in a large clinical trial.74 Thus, despite the large body of experimental and biomarker data supporting a role for oxidized LDL in the pathogenesis of atherothrombosis, no antioxidant intervention has thus far proven successful in limiting cardiovascular risk.
Other specific oxidant pathways may also contribute to atherothrombosis. Inflammatory cells associated with plaques, notably a subpopulation of macrophages in atheromata, can contain the enzyme myeloperoxidase (MPO), which can generate the potent oxidant species hypochlorous acid (HOCl).75,76 HOCl can mediate chlorination of tyrosyl residues in proteins. Indeed, the products of HOCl modification accumulate in plaques, indicating the action of MPO in situ in human lesions. In particular, the modification of a specific tyrosyl residue in apolipoprotein A-I can impede reverse cholesterol transport mediated by HDL.77,78
MPO constitutes a considerable portion of the protein content of granulocytes. Although sparse in undisrupted atherosclerotic lesions, arterial thrombi can trap granulocytes. Thus, MPO-generated HOCl levels may increase selectively at sites of fresh thrombosis. The pathophysiologic implications of this local oxidative stress in aggravating the consequences of arterial thrombosis remain poorly explored.
One of the drivers of hypertension, angiotensin II, not only acts as an arterial vasoconstrictor, but also augments oxidative stress. In particular, angiotensin II can increase the activity of NAD(P)H oxidases in artery wall cells, notably smooth muscle.79 These oxidases produce superoxide anion, a potential contributor to local oxidative stress in arterial plaques. Whether some of the clinical benefit ascribed to interventions that limit angiotensin II production or signaling arises from reduced superoxide anion production remains speculative.
Infection in Atherosclerosis
Among emerging risk factors, infectious agents, both viral and bacterial, have received periodic attention as promoters of atherogenesis through the decades. The seroepidemiologic associations between infectious agents and atherosclerosis have proved subject to considerable confusion.80,81,82 The more recent well-powered and appropriately adjusted seroepidemiologic surveys show little or no consistency in the association of infectious agents with atherothrombotic events. Some have advocated a relationship between burden of exposure to infectious agents and atherosclerotic risk, rather than infection with a specific etiologic virus or microbe.81
In situ studies have localized infectious agents or their products to many human atherosclerotic plaques.83 Whether these invaders or their products promote the disease process remains unproven. In particular, some well-powered intervention trials with antimicrobial agents, including macrolides and a fluoroquinolone, have not shown reduction in recurrent cardiovascular events in individuals who have known coronary heart disease.84
While infectious agents may not promote atherogenesis through local actions in the plaque itself, systemic responses to infection could quite reasonably contribute to heightened risk for atherothrombotic events. For example, bacterial products, through ligation of pattern recognition receptors including members of the toll-like receptor family, can augment the expression of proinflammatory cytokines.85 Ultimately, cytokines produced in response to bacterial products including endotoxins can substantially augment circulating levels of interleukin-6 (IL-6). IL-6 regulates the acute-phase response of hepatocytes and markedly increases production by the liver of fibrinogen and the endogenous inhibitor of fibrinolysis— plasminogen activator inhibitor-1 (PAI-1) (FIGURE 89.3). Thus, infectious agents can tip the homeostatic-hemostatic balance, from one that favors fibrinolysis and limits thrombosis to a pathologic situation characterized by enhanced thrombotic potential and reduced fibrinolytic capacity. These changes
pertain not only to the “fluid phase” of blood, but also locally at the level of the plaque (FIGURE 89.4). Several cell types in the atherosclerotic plaque can also increase the local production of PAI-1, a function that can promote thrombus accumulation. So infectious agents, even if not driving pathogenesis of atherosclerosis per se, can augment the likelihood of thrombotic complications of this disease secondarily.
pertain not only to the “fluid phase” of blood, but also locally at the level of the plaque (FIGURE 89.4). Several cell types in the atherosclerotic plaque can also increase the local production of PAI-1, a function that can promote thrombus accumulation. So infectious agents, even if not driving pathogenesis of atherosclerosis per se, can augment the likelihood of thrombotic complications of this disease secondarily.
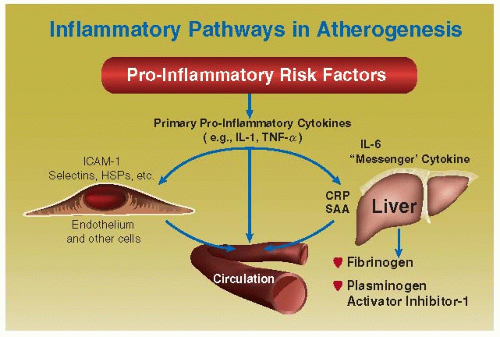 FIGURE 89.3 Pathways by which vascular and extravascular sources of inflammation result in circulating levels of serum markers that provide a reflection of the underlying inflammatory response. Systemic or local inflammation, either in the blood vessel itself or elsewhere, triggers the production of multipotent proinflammatory cytokines, denoted here as “primary” (e.g., IL-1β or TNF-α). These primary cytokines can directly elicit production by endothelial and other cells of adhesion molecules, procoagulants, and other mediators that may be released in soluble form into circulating blood. Primary cytokines also stimulate the production of the “messenger” cytokine IL-6, which induces expression of hepatic genes encoding acute-phase reactants found in blood, including CRP and serum amyloid-A (SAA). These markers in serum therefore provide a window on the inflammatory status of the individual, otherwise inaccessible in the intact subject. (From Libby P, Ridker PM. Novel inflammatory markers of coronary risk: theory versus practice. Circulation 1999;100:1148-1150, with permission.) |
Inflammation in the Pathogenesis of Atherosclerosis
Considerable current information implicates inflammation as a major participant in the pathogenesis of atherosclerosis and its complications. In particular, many of the traditional risk factors may produce their atherogenic effects by harnessing proinflammatory pathways ordinarily involved in host defenses (FIGURE 89.3). For example, oxidized phospholipids associated with modified LDL particles can augment inflammatory functions in vascular wall cells and macrophages. Angiotensin II can increase levels of proinflammatory mediators, including leukocyte adhesion molecules (such as vascular cell adhesion molecule-1) and proinflammatory cytokines (including IL-6). Thrombin itself not only generates fibrin, but through interaction with protease-activated receptors, also can elicit proinflammatory functions from cells implicated in atherogenesis.
Indeed, platelets not only participate in clot formation, but their granular constituents also include proinflammatory mediators, including RANTES and CD40 ligand (CD154) (FIGURE 89.5). Thus inflammation and thrombosis beget one another, inextricably linking these two branches of host defenses. Numerous experimental studies conducted in cultured cells and in animals, such as genetically altered mice, assert a causal role for inflammatory mediators in the aggravation of atherosclerosis.86 Biomarker studies in humans support the relevance of these laboratory experiments to the human disease, as discussed in detail below. Of emerging biomarkers, those reflecting inflammation have come most rapidly into clinical practice, as discussed later in this chapter.
INITIATION OF ATHEROSCLEROTIC LESIONS
As noted earlier, the very first steps in the evolution of atheroma may be the only aspects of this disease that do not involve thrombosis. Atherosclerosis usually develops in the context of elevated risk factors. An altered pattern of plasma lipoproteins is a well-established risk factor for lesion initiation.
We have learned much about the earlier stages of atherogenesis by studying experimental animals, which develop atheromatous lesions when they consume diets enriched with cholesterol and saturated fat. Such animal experiments likely represent a caricature of human atherosclerosis, because the achieved levels of lipoproteins exceed by far those found in most human patients with atherosclerosis. Despite the convenience of hypercholesterolemic models of atherogenesis, many other risk factors contribute to this disease. Nonetheless, these wellstudied experimental preparations have afforded considerable insight into one pathway for lesion initiation.
Extracellular lipid tends to accumulate in the intima when high levels of plasma lipoproteins bearing cholesterol circulate in plasma (FIGURE 89.6).87 Part of this accumulation may result from increased permeability of the endothelial layer to lipoproteins at sites of lesion predilection,88,89 undoubtedly related to hydrodynamic changes at these locations, which typically localize
near branch or flow dividers in the arterial tree. In addition to increased penetration into the artery wall, increased retention of lipoprotein particles can contribute to accumulation of lipid.90 Whatever causes them to accumulate in the intima, lipoproteins provoke biologic reactions that seem to initiate atherogenesis.91 Lipoprotein particles undergo modification in the arterial intima.92 In particular, binding of lipoproteins to proteoglycan in the intima may both retard the egress of these particles and render them more susceptible to modification by oxidation.93,94 These findings have stimulated the formulation of a “responseto-retention” model for the initiation of atherosclerosis.95 Glycation in the presence of hyperglycemia represents another form of modification of LDL particles in the intima.96
near branch or flow dividers in the arterial tree. In addition to increased penetration into the artery wall, increased retention of lipoprotein particles can contribute to accumulation of lipid.90 Whatever causes them to accumulate in the intima, lipoproteins provoke biologic reactions that seem to initiate atherogenesis.91 Lipoprotein particles undergo modification in the arterial intima.92 In particular, binding of lipoproteins to proteoglycan in the intima may both retard the egress of these particles and render them more susceptible to modification by oxidation.93,94 These findings have stimulated the formulation of a “responseto-retention” model for the initiation of atherosclerosis.95 Glycation in the presence of hyperglycemia represents another form of modification of LDL particles in the intima.96
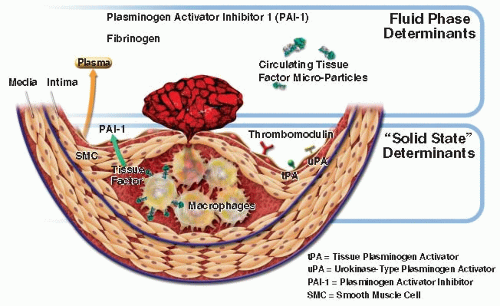 FIGURE 89.4 A two-state model of atherothrombosis. The high-risk atheroma has a thin fibrous cap overlying a large lipid core that contains tissue factor-bearing macrophages. When the fibrous cap fractures, coagulation proteins in the fluid phase of blood gain access to tissue factor-associated macrophages and tissue factor-bearing microparticles derived from apoptotic cells in the solid state of the plaque. These events trigger thrombus formation on the ruptured plaque. The clinical consequences depend on the amount of tissue factor and apoptosis in the plaque’s core and on the levels of fibrinogen and PAI-1 in the fluid phase of blood. The interaction of the fluid phase with the solid state will determine whether a given plaque disruption provokes a partial or transient coronary artery occlusion (that can be clinically silent or, less commonly, cause an episode of unstable angina) or a devastating persistent and occlusive thrombus that can precipitate an acute MI. Inflammation regulates the thrombotic/fibrinolytic balance in both the solid state and the fluid phase, as PAI-1 and fibrinogen are both acute-phase reactants and because the inflammatory mediator CD40 ligand (CD154) induces tissue factor expression. EC, endothelial cell; PAI-1, plasminogen activator inhibitor 1; SMC, smooth muscle cell; tPA, tissue plasminogen activator; uPA, urokinase-type plasminogen activator. (From Libby P, Theroux P. Pathophysiology of coronary artery disease. Circulation 2005;111:3481-3488, with permission.) |
Modified lipoproteins can elicit inflammatory mediators, such as proinflammatory cytokines, from the resident cell types in the normal artery: ECs and SMCs.97,98,99 Such cytokines, protein mediators of inflammation, can elicit the expression of leukocyte adhesion molecules on the surface of ECs. Leukocytes attached to such “activated” ECs at sites of lesion predilection can enter the intima by diapedesis between EC junctions.100,101 Chemotactic cytokines, such as macrophage chemoattractant protein-1 (MCP-1) and other chemokines, probably cause such directed migration of leukocytes. Atheromata overexpress these mediators, perhaps partly in response to modified lipoprotein particles. Once resident in the intima, mononuclear phagocytes can imbibe the modified lipoproteins that have accumulated extracellularly. The phagocytic cells bind and internalize these modified lipoprotein particles using “scavenger receptors.”102 The lipid-laden macrophage so produced constitutes the foam cell—the hallmark of the initial lesion of atherosclerosis, the fatty streak (FIGURE 89.6).
We owe much of our knowledge about the initiation of atherosclerosis to studies in well-defined animal preparations. But none of the available animal preparations mimic human atherosclerosis perfectly, not even the power offered by genetically altered mice (discussed later). Hence, the concurrent study of human lesions has proven an important complement to the animal work. With regard to the initiation of lesions, some human studies have furnished insight. Autopsy studies on young Americans, including those killed in armed conflicts in the 1950s and 1960s, showed a high prevalence of fatty streaks and even raised lesions in coronary arteries.103,104 Systematic study of arteries collected from Americans younger than 35 years and dying of noncardiac causes has affirmed the high prevalence of nascent atheroma in humans in the third and fourth decades of life.105,106 The general pattern of disease morphology agrees with the inferences made by extrapolation from atherosclerotic animals.
Related posts:
 The Field of Hemostasis and Thrombosis: Selected Translational Achievements
The Field of Hemostasis and Thrombosis: Selected Translational Achievements
 Integrín αIIbβ3 and Platelet Aggregation
Integrín αIIbβ3 and Platelet Aggregation
 Coagulation-Independent Signaling of the Extrinsic Coagulation Pathway
Coagulation-Independent Signaling of the Extrinsic Coagulation Pathway
 Inherited Thrombocytopenias
Inherited Thrombocytopenias
 Pathogenesis and Treatment of Biomaterial-Associated Thrombosis
Pathogenesis and Treatment of Biomaterial-Associated Thrombosis
 Blood Management in the Cardiovascular Surgical Patient
Blood Management in the Cardiovascular Surgical Patient
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


