Pathophysiology and Vascular Biology of Atherosclerosis
Subroto Chatterjee
Over 50 years ago, Dr. Alfred Blalock, a cardiovascular surgeon at the Johns Hopkins Hospital commented, “The heart is an innocent bystander in coronary heart disease; the real culprit is the tubing that supplies blood to the heart muscles.” This chapter discusses historical and recent evidence that confirms and extends the importance of the vasculature in the pathophysiology of atherosclerosis. Two major and interrelated pathways have been proposed to explain the pathophysiology and vascular biology of atherosclerosis: (i) the oxidation of low-density lipoproteins (LDL) pathway and (ii) the inflammatory pathway. Both pathways manifest several key events that can contribute to the initiation and progression of atherosclerosis.
The pathophysiology of atherosclerosis involves the transition of a normal blood vessel into an atherosclerotic blood vessel (Fig. 4.1). Activation/injury to the endothelium, uptake and retention of LDL and its oxidation, infiltration of monocytes and their conversion to macrophages that become lipid-laden foam cells, arterial smooth muscle cell (ASMC) proliferation from the tunica media and migration to the intima, and thrombosis constitute the five major characteristics in atherogenesis. Of note, a number of cardiovascular disease (CVD) risk factors such as elevated LDL, hypertension, insulin resistance, and cigarette smoking promote endothelial dysfunction and a slow but progressive thickening of the arterial intima.
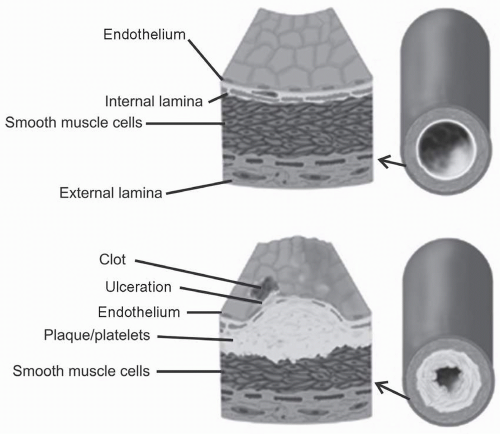 FIGURE 4.1 Diagrammatic representation of the pathophysiology of atherosclerosis and plaque formation. Note the focal accumulation of lipid-laden foam cells beneath an intact arterial endothelial lining. (Reproduced with permission from Your Surgery.com. Arterial Surgery of the Leg. http://www.yoursurgery.com/ProcedureList.cfm) |
THEORETICAL CONSIDERATIONS
Pathway 1: Oxidation of LDL and Its Consequences on Atherosclerosis
Importance of LDL Modification and Oxidation
The oxidation of LDL (ox-LDL) is one of the major contributing factors to the activation of the vascular endothelium. Brown and Goldstein (1) showed that macrophages express few LDL receptors; however, LDL reacted with acetic anhydride in
vitro to form acetyl-LDL that is avidly taken up by macrophages by receptor-mediated endocytosis and can lead to excessive cholesterol uptake and the conversion of the macrophage into a foam cell. Acetylation of LDL removes positive charges from the epsilon amino groups of lysine and converts a weakly anionic lipoprotein into a strongly anionic molecule (1). As originally shown by Fogelman and colleagues, malondialdehyde can also react with the lysine residues of LDL and convert LDL to a form recognized with high affinity by macrophages (2). Pioneering studies by Chisholm and colleagues (3) showed that LDL cytotoxicity could be induced by free radical peroxidation of lipid.
vitro to form acetyl-LDL that is avidly taken up by macrophages by receptor-mediated endocytosis and can lead to excessive cholesterol uptake and the conversion of the macrophage into a foam cell. Acetylation of LDL removes positive charges from the epsilon amino groups of lysine and converts a weakly anionic lipoprotein into a strongly anionic molecule (1). As originally shown by Fogelman and colleagues, malondialdehyde can also react with the lysine residues of LDL and convert LDL to a form recognized with high affinity by macrophages (2). Pioneering studies by Chisholm and colleagues (3) showed that LDL cytotoxicity could be induced by free radical peroxidation of lipid.
Work in Daniel Steinberg’s laboratory showed that cultured endothelial cells (ECs) could convert human LDL into a form recognized by the acetyl-LDL receptor (4). Subsequently, this group found that an EC-derived chemotactic activity for mouse peritoneal macrophages could be inhibited either by addition of EC-modified LDL or by LDL oxidized chemically in vitro (4). EC-modified LDL was shown previously to contain peroxidized lipids, increased lysophosphatidylcholine (lysoPC), and partially degraded apolipoprotein B (apoB) (see also the following text). Inhibitory activity was present in the lipid extract of EC-modified LDL but not in that of native LDL, representing peroxidized lipid components. Because EC-modified LDL is taken up in part by way of the acetyl-LDL receptor, the effects of acetyl-LDL were tested. Rather than inhibiting chemotaxis, acetyl-LDL showed intrinsic positive chemotactic activity. These studies suggested that ox-LDL may recruit macrophages into the subendothelial space and these cells then become “trapped” there because of the inhibitory effects of peroxidized lipid components in modified forms.
In Vitro and In Vivo Ox-LDL
The cholesterol (or LDL) hypothesis is (i) the higher the LDL levels, the greater the prevalence of CVD, and (ii) lowering LDL decreases risk of CVD. Ox-LDL (or modified by glycation) plays a pivotal role in the development of atherosclerosis and CVD. The fundamental premise was that macrophages do not have LDL receptors but do express various receptors/scavenger receptors that can take up modified/ox-LDL, thus contributing to foam cell formation and atherogenesis (1,2,3,4,5,6,7,8).
In Vitro Ox-LDL. Ox-LDL can be achieved by dialysis against a solution of FeSO4 to yield minimally modified ox-LDL (MM-LDL), which has undergone antioxidant depletion and oxidation of arachidonic acid-containing phospholipids, much less linoleic acid oxidation, with little or no protein modification (8). Under relatively more rigorous conditions, for example, dialysis against Cu2SO4, LDL can be fully oxidized including apoB and a variety of lipids. Also when LDL is exposed to vascular cells grown in tissue culture such as ECs and ASMCs, the LDL is modified and manifests altered physical-chemical properties, for example, increase in electrophoretic migration and oxidized products detectable by thio-barbituric acid assay and specific antibodies.
In Vivo Ox-LDL
Such ox-LDL requires an interaction with erythrocytes (hemoglobin) that are plentiful in the circulating blood. Hemoglobin is released during bleeding episodes. Also blood can enter the vascular wall by the rupture of the vasa vasorum, intraplaque hemorrhaging, and myocardial infarction (10) (see also Chapter 3). Previous studies have shown that under hypoxic conditions the majority of hemoglobin is in double and triple E liganded states, which can oxidize proteins much more effectively and rapidly than oxyhemoglobin. Thus, erythrocytes not only contribute to a source of hemoglobin with which LDL could react, but also donate a significant amount of cholesterol due to its desquamation and uptake by the macrophages. To test the role of hemoglobin in LDL modification and subsequent effects on phenotypic changes, such as proliferation of ASMCs, my colleagues and I conducted the following experiments. LDL was oxidized with oxygenated normoxic hemoglobin, partially oxygenated hypoxic hemoglobin, and fully oxygenated hemoglobin; Cu2SO4-modified LDL served as a positive control. We observed that all such modified LDL stimulated the phosphorylation of p44 mitogen-activated protein kinase (MAPK) via activating this enzyme. And this was accompanied by a marked, concentrationdependent increase in ASMC proliferation (11).
Inflammation, restenosis, and ulcerations are several conditions in vivo that may contribute to erythrocyte lysis, release of hemoglobin, and its oxidation in blood (see also Chapter 3). The subsequent interaction of oxidized hemoglobin or methemoglobin with LDL may well contribute to atherogenesisstimulating ASMC proliferation and cholesterol uptake by macrophages and ASMCs and their deposition under the vascular endothelium (Fig. 4.1).
Components of MM-LDL and their Effect on Phenotypic Changes Contributing to Atherogenesis
Since atherosclerosis is a disease of the vessel wall involving the accumulation of cholesterol, chronic infection, cell proliferation, cell death, and thrombus formation, this section examines how compounds present in modified LDL can participate in the process of atherosclerosis.
Oxidized Phospholipids. The chemical structures of various compounds found in MM-LDL are shown in Figure 4.2. Ox-PAPC (1-palmitoyl-2-arachidonyl-sn-glycero-3-phosphorylcholine) as well as its components include 1-palmitoyl-2-(5-oxovaleroyl)-sn-glycero-3-phosphocholine (POVPC), 1-palmitoyl-2-glutrayl-sn-glycero-3-phosphatidylcholine (PGPC),1-palmitoyl-2-2-isopentanoyl-sn-glycero-3-3-phoshocholine (PEIPC), and epoxycyclopentenone (PECPC).
Studies in vitro in human arterial ECs showed that the expression of monocyte chemoattractant protein (MCP) was stimulated by oxidized phospholipids facilitating the adhesion of circulating monocytes to the endothelium, and thereby contributing to the initiation of atherosclerosis (9). There is a structure/activity relationship among these oxidized phospholipids in regard to their stimulation of the proliferation of ASMCs. For example, only POVPC exerted a concentrationdependent stimulation in ASMC proliferation by activating an “oxygen-sensitive signaling pathway” (Fig. 4.3). The various components of this pathway include activation of LacCer synthase (LCS) to generate LacCer; LacCer serves as a sphingolipid second messenger promoting superoxide generation; kinase activation of, for example, p44 MAPK phosphorylation; c-fos expression; and cyclin-1 expression, leading to proliferation of ASMCs (Fig. 4.3). This phenotypic change in the ASMC
can be mitigated by a molecule that is an analog of ceramide and inhibits both glucosylceramide synthase and LCS, namely D-threo-1-phenyl-2-decanoylamino-3-morpholino-1-propanol (D-PDMP) (12).
can be mitigated by a molecule that is an analog of ceramide and inhibits both glucosylceramide synthase and LCS, namely D-threo-1-phenyl-2-decanoylamino-3-morpholino-1-propanol (D-PDMP) (12).
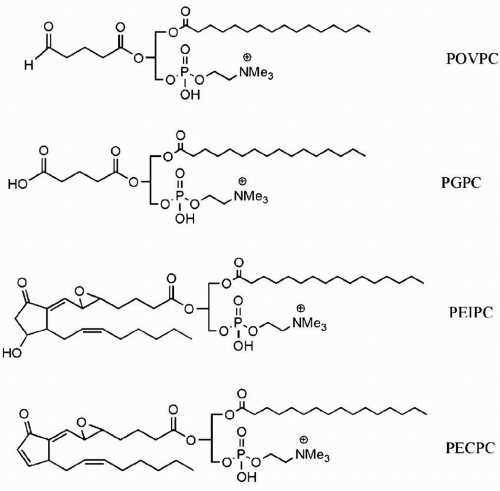 FIGURE 4.2 Chemical structures of oxidized phospholipids found in minimally modified LDL. LDL, low-density lipoproteins. (Presented with permission of Dr. Judy Berliner.) |
The presence of POVPC, PGPC, and PEIPC in human arterial lesions suggested that these oxidized phospholipids had a pathophysiologic role in atherosclerosis. For example, oxidized phospholipids modulate the thrombotic burden by decreasing thrombomodulin, and by increasing tissue factor, the secretion of tissue factor pathway inhibitor, and plasminogen activator inhibitor (13). Furthermore, ox-PAPC inhibited lipopolysaccharide (LPS)-induced expression of interleukin-8 (IL-8), depleted cholesterol, and increased the expression of sterol regulatory element binding protein (SREBP), thus promoting LDL receptor transcription (13). In addition, ox-PAPC was shown to stimulate the activity of a cell membrane-bound neutral sphingomyelinase (N-SMase), increase ceramide production (the catabolic product of N-SMase action), and inhibit LPS-induced IL-8 generation. This process was blocked by GW-4869, an inhibitor of N-SMase (14).
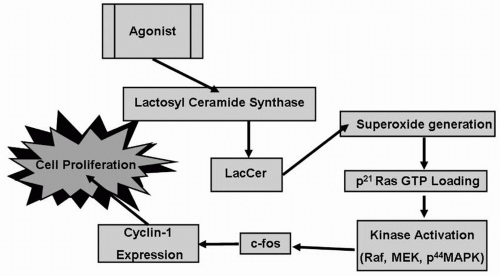 FIGURE 4.3 Agonists such as PDGF, oxidized LDL, and POVPC-induced ASMC proliferation recruit lactosyl ceramide synthase to generate LacCer. In turn, LacCer activates an “oxygen-sensitive signaling pathway” triggered by the generation of superoxides and contributes to cell proliferation. PDGF, platelet-derived growth factor; LDL, low-density lipoproteins; POVPC, 1-palmitoyl-2-(5-oxovaleroyl)-sn-glycero-3-phosphocholine; ASMC, arterial smooth muscle cell; LCS, lactosyl ceramide synthase; LacCer, lactosyl ceramide. |
In aggregate, these findings indicate that oxidized phospholipids (derived from MM-LDL) are key players in atherogenesis through the activation of multiple signaling pathways, which contribute to phenotypic changes such as cell adhesion, migration, proliferation, and thrombosis.
Components of Fully Oxidized LDL and their Effects on Phenotypic Changes Contributing to Atherosclerosis
LDL oxidation in vitro results in a decrease in the content of cholesterol and cholesteryl ester and a rise in oxysterols. For example, during the early stages of oxidation with Cu2SO4, 7-hydroperoxy cholesterol (7-OOH) is an important intermediate of cholesterol oxidation, and 7-ketocholesterol (7-OH) is the bulk of oxidized form of cholesterol. It plays an important role in inhibiting apolipoprotein A-I (apoA-I)-induced reverse cholesterol efflux (see the following text). Also in oxidized LDL, cholesteryl insolate is converted to 7-ketocholesteryl-9-carboxynonanoate (Fig. 4.4). The important feature of this and other compounds shown in Figure 4.4 is that they serve as ligands for β2-glycoprotein (β2-GPI) (see the following text). In contrast, 7-ketocholesteryl-12-carboxy(keto) dodecanoate is the major oxidized cholesterol derivative in heavily oxidized LDL (Fig. 4.4).
Adverse Effects of Oxidation on HDL and Reverse Cholesterol Transport. ApoA-I, the major protein of HDL facilitates the efflux of cholesterol from macrophages in foam cells, a process felt to promote reverse cholesterol transport and decrease the cholesterol burden of macrophages (see also Chapter 9). This antiatherosclerotic process is inhibited by 7-ketocholesterol. Ox-LDL also results in the modification of the steryl ester fatty acyl groups. For example, cholesteryl linoleate is converted to 9-oxo-nanoyl cholesterol; cholesteryl-hydroperoxy-octadecanoate and cholesteryl octadecadienoate were also found as major
products in ox-LDL. Further studies reveal that cholesteryl-hydroperoxy-octadecanoate inhibits platelet-derived growth factor (PDGF). Also oxidized linoleic acid/arachidonic acid derivatives such as 9-hydroxydecadienoic acid, 13-hydroxydecadienoic acid, and 15-hydroxyeicosatetraenoic acid were found to bind to peroxisome proliferator-activated receptors and activate PPAR-γ. In contrast, oxidized phospholipids from LDL bind to PPAR-α. These in turn bind to heterodimers with retinoid receptors to PPAR response elements in the promoter region of target genes that alter lipid and glucose metabolism, which can lead to lipodystrophy, obesity, the metabolic syndrome, and diabetes.
products in ox-LDL. Further studies reveal that cholesteryl-hydroperoxy-octadecanoate inhibits platelet-derived growth factor (PDGF). Also oxidized linoleic acid/arachidonic acid derivatives such as 9-hydroxydecadienoic acid, 13-hydroxydecadienoic acid, and 15-hydroxyeicosatetraenoic acid were found to bind to peroxisome proliferator-activated receptors and activate PPAR-γ. In contrast, oxidized phospholipids from LDL bind to PPAR-α. These in turn bind to heterodimers with retinoid receptors to PPAR response elements in the promoter region of target genes that alter lipid and glucose metabolism, which can lead to lipodystrophy, obesity, the metabolic syndrome, and diabetes.
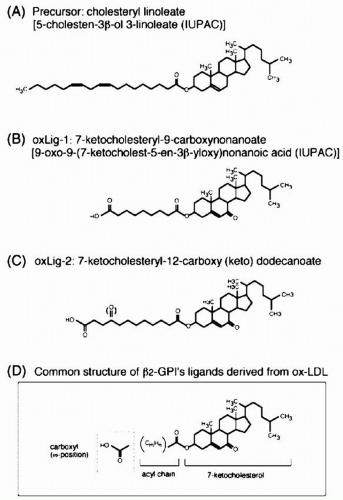 FIGURE 4.4 Chemical structures of oxidized lipids in ox-LDL. Ox-LDL, oxidation of LDL. (Reproduced with permission from Matsuura E, Hughes GR, Khamashta MA. Oxidation of LDL and its clinical implication. Autoimmun Rev. 2008;7:558-566.) |
Ox-LDL can also contain several short-chain phosphatidylcholines, as the long-chain fatty acids (C18-20 polyunsaturated fatty acids) break away from the parent molecule. For example, if linoleic acid containing PC is oxidized, it produces 9-CHO phosphatidylcholine or 9-COOH phosphatidylcholine. In fact, an antibody against 9-CHO phosphatidylcholine has been useful in detecting these epitopes in foam cells in atherosclerotic lesions (15).
Role of Receptors for Ox-LDL in Atherosclerosis
Macrophages predominantly express various classes of ox-LDL receptors collectively called scavenger receptors. However, lectin-like ox-LDL receptor-1 (LOX-1) is expressed not only in macrophages, but also in activated human vascular ECs and ASMCs (16). Interestingly, ox-LDL, the ligand for the LOX-1 receptor, as well as several proinflammatory cytokines can increase LOX-1 expression. The binding of ox-LDL to LOX-1 raises the level of reactive oxygen species (ROS), reduces the level of nitric oxide, and activates nuclear factor κB (NF-κB) and MCP, which in turn facilitates the adhesion of circulating monocytes to the endothelium. Additional consequences of ox-LDL and LOX-1 interaction include an induction of apoptosis involving Bcl-2, cIAP-2, release of cytochrome c from the mitochondria, and the activation of several caspases (17). In human ASMC, we observed that ox-LDL at high concentrations (100 µg/mL) can stimulate the activity of N-SMase, generate ceramide, release cytochrome c from mitochondria, and induce marked apoptosis (18).
Inflammation and Atherosclerosis
Role of Inflammation in the Onset of Atherosclerosis
Because populations of humans where the total cholesterol is <150 mg/dL and the LDL-C is <80 mg/dL get very little CVD, it has been proposed that elevated LDL is most likely the “sine qua non” of atherosclerosis. However, there are a myriad of other reactions that occur beyond LDL oxidation, and inflammation is a paramount component of the atherosclerotic process. Two hypotheses have been proposed: (i) inflammation primarily results from the pathophysiologic effects of ox-LDL, and (ii) inflammation contributes to the atherosclerotic process independent of LDL (6).
Other Inflammatory Molecules. Primary proinflammatory cytokines such as tumor necrosis factor alpha (TNF-α), secreted by tissues in response to a bacterial infection, can activate the vascular endothelium by releasing ROS, growth factors, cell adhesion molecules, and matrix metalloproteinases (MMPs). These molecules partake in phenotypic changes such as cell proliferation, adhesion, migration, angiogenesis, and apoptosis of the cells of the vascular wall. Such events may well predict the initiation, progression, and severity of atherosclerosis and plaque rupture. But our knowledge of the application of these biomarkers in atherosclerosis as valid predictors of CVD is limited and requires further evaluation (6). Additional cytokines are also produced upon interaction of cells of the vascular wall with TNF-α. One such example is the generation of a messenger cytokine, namely, interleukin-6 (IL-6) by ASMC and the secretion of IL-6 into the bloodstream. In turn, IL-6 binds to liver cells and stimulates the generation of acute phase reactants such as C-reactive protein (CRP) (see also section “Practical Considerations”). There is also growing evidence that the generation of a small molecular weight sphingolipid/LacCer may be a common feature involved in mediation of atherosclerosis by ox-LDL and inflammation/TNF-α (see the following text). Hence, drugs targeting this molecule may be useful in preventing/interfering with atherosclerosis.
Although one of the important events in the pathogenesis of atherosclerosis is the focal accumulation of lipid-laden foam cells beneath an intact arterial endothelial lining (Fig. 4.1),
localized attachment of circulating monocytes and lymphocytes to the arterial endothelium precedes the formation of early foam cell lesions (19). Studies showed that other risk factors such as cigarette smoking, high blood pressure, diabetes, and bacterial infection all contributed to atherosclerosis by eliciting inflammation in vascular tissues (20,21,22).
localized attachment of circulating monocytes and lymphocytes to the arterial endothelium precedes the formation of early foam cell lesions (19). Studies showed that other risk factors such as cigarette smoking, high blood pressure, diabetes, and bacterial infection all contributed to atherosclerosis by eliciting inflammation in vascular tissues (20,21,22).
Related posts:
 Use of Noninvasive Methods to Diagnose Cardiovascular Disease in Dyslipidemic Patients
Use of Noninvasive Methods to Diagnose Cardiovascular Disease in Dyslipidemic Patients
 Dyslipidemia in Children and Adolescents
Dyslipidemia in Children and Adolescents
 Dietary Treatment of Dyslipidemia
Dietary Treatment of Dyslipidemia
 Disorders of Hypertriglyceridemia
Disorders of Hypertriglyceridemia
 Dyslipidemia in the Elderly: Special Insights to Inform the Management Strategy
Dyslipidemia in the Elderly: Special Insights to Inform the Management Strategy
 Trans Fatty Acids, Dyslipidemia, and Cardiovascular Disease
Trans Fatty Acids, Dyslipidemia, and Cardiovascular Disease
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


