Disorders of Hypertriglyceridemia
Michael A. Miller
In 1959, Drs. Albrink and Mann reported the first association between elevated triglycerides (TG) and cardiovascular disease (CVD) (1). Subsequent case-control and longitudinal studies affirmed that high TG were prevalent in patients with pre-existing CVD and predictive of initial CVD events. Yet, despite the epidemiologic data supporting TG as a risk factor for CVD, its association was consistently weakened after adjustment for other covariates, most notably high-density lipoprotein cholesterol (HDL-C) (2). Despite National Institutes of Health (NIH) consensus conferences and several decades of research, the importance of TG in CVD risk assessment remains controversial and unresolved. What is generally accepted is that elevated levels of circulating TG are common in societies with high rates of obesity and diabetes mellitus. Unfortunately, the impressive strides made in reducing population-based CVD risk factors such as cigarette smoking in the United States have been offset by the reciprocal increases observed in visceral obesity, insulin resistance, and the metabolic syndrome, all of which are accompanied by hypertriglyceridemia.
This chapter focuses on disorders of TG metabolism and includes a review of the biochemistry, metabolism, and pathophysiology of TG-rich lipoproteins, proposed mechanisms relating hypertriglyceridemia to elevated CVD risk, and genetic abnormalities associated with abnormal TG. The evaluation of TG as a biomarker versus CVD risk factor will be followed by a review of clinical trials evaluating TG-lowering therapies on CVD risk. The final section will identify and discuss unanswered questions and ideas for future areas of scientific and clinical investigation.
THEORETICAL CONSIDERATIONS
Fundamental Biochemistry, Metabolism, and Pathophysiology of TG-rich Lipoproteins
Dietary Fat Utilization
Originating from dietary (exogenous) and hepatic (endogenous) sources, TG provide the fuel for energy utilization and storage in mammalian tissues. In the exogenous pathway, dietary TG are digested by pancreatic lipase and subsequently mixed with bile. The emulsified fat particles are hydrolyzed in the lower duodenum by pancreatic and intestinal lipases. Short- and medium-chain fatty acids are directed to the portal circulation, whereas long-chain fatty acids are processed into chylomicrons (CM) via intestinal microvilli incorporation. Although approximately 50% of dietary cholesterol is reabsorbed by the microvilli of intestinal epithelial cells, virtually all long-chain fatty acids are absorbed and re-esterified into TG under normal physiologic conditions. Free fatty acids (FFA) and monoacylglycerides (MAG) traverse the jejunal brush border membrane and are transported to the endoplasmic reticulum by a fatty acid-binding protein (FABP) (Fig. 7.1). Intracellular re-esterification of TG is mediated by interactions between acyl-CoA monoacylglycerol acyltransferase (MGAT), acyl-CoA diacylglycerol acyltransferase (DGAT), and microsomal triglyceride transfer protein (MTP), the lipidation of apolipoprotein B-48 (apoB-48) and subsequent formation of prechylomicron particles, with apolipoprotein A-IV (apoA-IV) serving as a cofactor. Liver FABP TG then directs budding of the pre-chylomicron transport vehicle (PCTV) where fusion of cis-Golgi occurs with coating protein II (COPII) and soluble N-ethylmaleimide sensitive factor attachment protein receptor (SNARE). In the Golgi, apolipoprotein A-I (apoA-I) is acquired and additional apoB-48 processing occurs before the release of chylomicrons into the lymphatic circulation (3). The mature chylomicron particle consists of a predominant TG core (˜95% of mass) with a small amount of cholesteryl ester. The outer surface of a chylomicron is composed of phospholipids (PL), free cholesterol, and apoB-48, apoA-I, and apoA-IV.
Hydrolysis of TG in Chylomicrons. Following release of secreted chylomicrons from the thoracic duct into the venous circulation, an exchange of apolipoproteins takes place with apolipoprotein E (apoE) and apolipoprotein C-II (apoC-II) (and to a smaller extent, apolipoprotein C-III [apoC-III]), replacing apoA-I and apoA-IV, the former of which is transferred to nascent HDL. As the chylomicron particle traverses through capillaries within systemic tissues such as heart, adipose, and muscle, it is partially hydrolyzed by lipoprotein lipase (LPL). Specifically, LPL attaches to the lumen of vascular endothelial cells by interacting with cell surface heparan-sulfate proteoglycans (HSPG). ApoE associates with HSPG, permitting anchoring of the chylomicron particle and facilitating apoC-II activation of LPL.
Barring genetic abnormalities in LPL or APOC2, the half-life (t½) of circulating chylomicrons approximates 5 to 10 minutes. In contrast, the t½ of circulating hepatically derived very-low-density lipoproteins (VLDL) is considerably longer
(i.e., 4 to 5 hours) due to the reduced surface area with which these smaller particles interact with LPL on the capillary or intra-arterial surface (4). If allelic or posttranslational defects produce functional defects in LPL or APOC2, chylomicrons and/or large VLDL particles will have an increased residence time in the circulation; the most severe cases of ensuing chylomicronemia may result in vessel branch occlusion or pancreatitis (see following text). Other conditions that do not favor optimal LPL-APOC2 hydrolytic interactions include increased production (or possibly reduced degradation) of apolipoprotein C-I (apoC-I) and apoC-III, which may displace apoE and result in reduced uptake of the lipoprotein particles by the hepatic LDL receptor-related protein (LRP) (5). Under physiologic conditions, the by-products of hydrolysis of TG as a consequence of LPL-APOC2 interactions are circulating fatty acids that are bound to albumin and transported as FFA to various tissues including myocardium and skeletal muscle for energy utilization or stored in adipose tissue.
(i.e., 4 to 5 hours) due to the reduced surface area with which these smaller particles interact with LPL on the capillary or intra-arterial surface (4). If allelic or posttranslational defects produce functional defects in LPL or APOC2, chylomicrons and/or large VLDL particles will have an increased residence time in the circulation; the most severe cases of ensuing chylomicronemia may result in vessel branch occlusion or pancreatitis (see following text). Other conditions that do not favor optimal LPL-APOC2 hydrolytic interactions include increased production (or possibly reduced degradation) of apolipoprotein C-I (apoC-I) and apoC-III, which may displace apoE and result in reduced uptake of the lipoprotein particles by the hepatic LDL receptor-related protein (LRP) (5). Under physiologic conditions, the by-products of hydrolysis of TG as a consequence of LPL-APOC2 interactions are circulating fatty acids that are bound to albumin and transported as FFA to various tissues including myocardium and skeletal muscle for energy utilization or stored in adipose tissue.
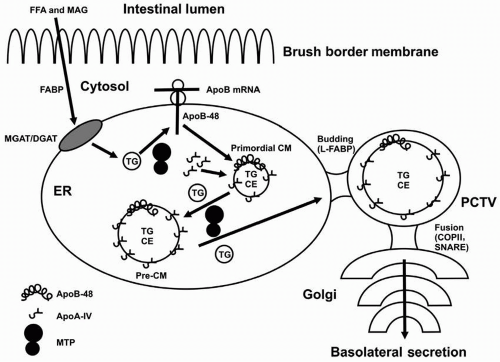 FIGURE 7.1 Assembly and secretion of CM. FFA and MAG are translocated to the ER by FABP whereupon TG are resynthesized by acyl-CoA: MGAT and acyl-CoA: DGAT. ApoB-48 is lipidated by MTP and the primordial CM are further lipidated to form pre-CM. The addition of apoA-IV permits additional lipidation, and L-FABP assists in budding of the PCTV and fusion via COPII and SNARE. The mature CM are secreted via the basolateral membrane. CM, chylomicrons; FFA, free fatty acids; MAG, monoacylglycerides; ER, endoplasmic reticulum; FABP, fatty acid-binding proteins; MGAT, monoacylglycerol acyltransferase; DGAT, diacylglycerol acyltransferase; apo, apolipoprotein; MTP, microsomal triglyceride transfer protein; L-FABP, liver fatty acid binding protein; PCTV, pre-CM transport vesicle; COPII, coating protein II; SNARE, soluble N-ethylmaleimide-sensitive factor attachment protein receptor. (Reproduced from Black DD. Development and physiological regulation of intestinal lipid absorption. I. Development of intestinal lipid absorption: cellular events in chylomicron assembly and secretion. Am J Physiol Gastrointest Liver Physiol. 2007;293: G519-G524, with permission.) |
Cellular Uptake of TG-rich Remnants. In addition to its pivotal catalytic role in hydrolyzing TG-rich lipoproteins (i.e., chylomicrons and VLDL), LPL also binds HSPG to form a nonenzymatic bridging interaction that facilitates the uptake of remnants by the hepatic LRP (6), an interaction opposed by apoC-III via inhibition of HSPG binding (7). Thus, the antiatherogenic roles attributable to LPL include hydrolyzing the TG-rich lipoproteins and serving as a ligand for remnant uptake by hepatic LRP receptors. While LPL is not expressed in adult liver, it is expressed in smooth muscle cells (SMC) (6) and macrophages (8), and macrophage LPL is expressed in atherosclerotic plaques (9) with higher activity reported in patients at increased risk of CVD including familial hypercholesterolemia (10) and diabetes mellitus (11). In addition to stimulating proinflammatory cytokine expression (i.e., tumor necrosis factor-α), macrophage LPL also interacts with HSPG to retain native and oxidized low-density lipoproteins (LDL) within the subendothelium (12). Taken together, LPL activation in adipose tissue appears to be antiatherogenic, whereas LPL produced by arterial wall macrophages promotes atherosclerosis, an effect that may be attenuated by inhibition of hydroxymethylglutaryl CoA reductase with statin therapy (13).
Atherogenicity of Remnant Lipoproteins
Although TG are not a significant component of atherosclerotic plaque, their downstream hydrolytic by-products, apoE-enriched remnant particles, have been isolated from atherosclerotic plaques and human aortic intima (14). Following
LPL-mediated hydrolysis of TG in chylomicrons and VLDL, the smaller, cholesterol-enriched remnant particles may be directly taken up by the endothelium in a manner believed to be analogous to LDL-mediated transcytosis (Fig. 7.2). Remnants induce vascular cell adhesion molecule expression (i.e., VCAM-1) and facilitate subendothelial uptake of monocytes where they are converted to macrophages. Macrophages activate T lymphocytes and cytokines that promote apoptosis, inflammation, and oxidation. Lipid engorgement converts macrophages into foam cells that develop into atherosclerotic plaques, whereas vascular SMC proliferate and migrate to the intima to form the fibrous cap (15).
LPL-mediated hydrolysis of TG in chylomicrons and VLDL, the smaller, cholesterol-enriched remnant particles may be directly taken up by the endothelium in a manner believed to be analogous to LDL-mediated transcytosis (Fig. 7.2). Remnants induce vascular cell adhesion molecule expression (i.e., VCAM-1) and facilitate subendothelial uptake of monocytes where they are converted to macrophages. Macrophages activate T lymphocytes and cytokines that promote apoptosis, inflammation, and oxidation. Lipid engorgement converts macrophages into foam cells that develop into atherosclerotic plaques, whereas vascular SMC proliferate and migrate to the intima to form the fibrous cap (15).
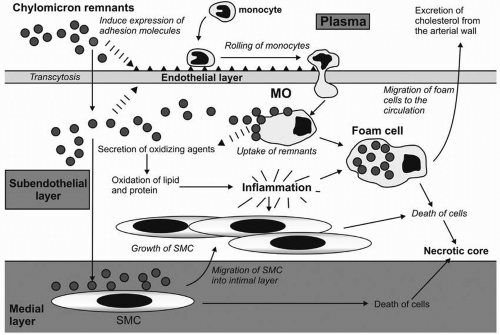 FIGURE 7.2 Chylomicron remnant-induced development of atherosclerosis. Chylomicron remnants traverse the endothelial layer by transcytosis and upregulate proatherothrombotic processes including adhesion molecule expression and monocyte migration into the subendothelial layer. The differentiated macrophages incorporate remnants in an unregulated manner and with modified LDL uptake, accelerate foam cell formation. Excessive remnant uptake may lead to secretion of oxygen reactive species and other toxins that result in cellular apoptosis. The migration of SMC to the subendothelial layer enables formation of the fibrous cap component of the atherosclerotic plaque. LDL, low-density lipoproteins; SMCs, smooth muscle cells. (Reproduced from Yu KC and Cooper AD. Postprandial lipoproteins and atherosclerosis. Front Biosci. 2001;6:D332-D354, with permission.) |
Proinflammatory Effects of TG-rich Lipoprotein. VLDL (including apoC-III-containing VLDL), as well as oxidized LDL, drive proinflammatory intracellular signaling pathways by interacting with the scavenger receptor and/or LRP, leading to activation of p38 mitogen-activated protein kinase and nuclear factor kappa beta (NF-κβ) (Fig. 7.3). This pathway results in the release of VCAM-1, facilitating recruitment of additional monocyte-macrophages and amplification of this process (16).
From a clinical standpoint, a high correlation exists between levels of TG and remnant lipoproteins (17), thereby providing proof of concept that the combination of high levels of both LDL-C and TG (as a surrogate for remnant lipoproteins) accentuate plaque formation and atherosclerotic progression (see also Proposed Mechanisms Relating Hypertriglyceridemia to Elevated CVD Risk).
Regulation of Adipocyte Lipolysis
Hormone-sensitive Lipase (HSL). While lipases (most notably, LPL) hydrolyze TG-rich lipoproteins for fat storage in adipocytes, the regulation of adipocyte lipolysis is governed by other proteins (Fig. 7.4). Specifically, catecholamines and insulin regulate lipolysis, with natriuretic peptides believed to contribute a smaller role. Catecholamines such as noradrenaline (norepinephrine) and adrenaline (epinephrine) bind to β-adrenoreceptors, principally β1 and β2, that couple with G proteins to stimulate adenylate cyclase. This in turn increases production of cyclic adenosine monophosphate (cAMP), activation of protein kinase A (PKA), and phosphorylation of HSL, thereby facilitating catecholamine-stimulated lipolysis. In contrast, insulin inhibits catecholamine-stimulated lipolysis. Uncoupling the Gi-receptor pathway results in inhibition of HSL, leading to decreased adipose tissue fatty acid mobilization and flux to the liver and culminating in lower hepatic secretion of VLDL and TG. Although nicotinic acid is mechanistically linked to these effects via direct inhibition of HSL, other compounds such as prostaglandin E2, neuropeptide Y, and adrenomedullin may also inhibit HSL through this pathway (18).
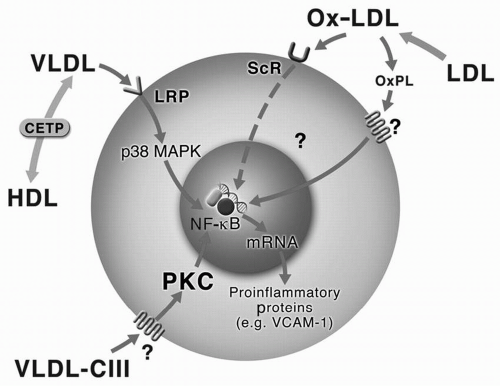 FIGURE 7.3. Proinflammatory effects of modified LDL and TG-rich lipoproteins. Ox-LDL promotes unregulated lipoprotein uptake by ScRs and associated OxPL activate inflammatory signaling pathways. TG-depleted VLDL remnants bind to LRP inciting downstream activation of p38 MAP kinase and NF-κB. ApoC-III-enrichment of VLDL activates PKC and stimulation of VCAM-1, thereby facilitating additional leukocyte (i.e., monocyte-macrophage) recruitment. Thus, mixed hyperlipidemia representing the combined effects of excess (modified) LDL and TG (as reflected by increased circulating remnant and apoC-III-enriched apoB) potentiates the atherosclerotic process. LDL, low-density lipoproteins; TG, triglycerides; Ox-LDL, oxidative modification of LDL; ScRs, scavenger receptors; OxPL, oxidized phospholipids; TG, triglycerides; VLDL, very-low-density lipoprotein; CETP, cholesteryl ester transfer protein; LRP, LDL-R-related proteins; NF-κB; nuclear factor kappa beta; apoC-III, apolipoprotein C-III; PKC, protein kinase C; VCAM-1, vascular cell adhesion molecule-1. (Reproduced from Libby P. Fat fuels the flame: triglyceride-rich lipoproteins and arterial inflammation. Circ Res. 2007;100:299-301, with permission.) |
Adipose Triglyceride Lipase (ATGL). The second important lipolytic enzyme in adipose tissue, ATGL, is the predominant lipase under basal conditions that hydrolyzes TG to diglycerides (Fig. 7.4). Although studies evaluating genetic variation of ATGL have identified structural polymorphisms that were associated with increased levels of FFA and fasting glucose, the association with body mass index and plasma TG have been considerably less pronounced (19).
Monoacylglycerol Lipase (MGL). A third enzyme, MGL, is not under hormonal control and hydrolyzes monoglycerides to FFA and glycerol (Fig. 7.4).
Natriuretic Peptides. In addition to catecholamines, atrial natriuretic peptide (ANP) and brain natriuretic peptide contribute to adipocyte lipolysis by activating natriuretic peptide receptor A, and, through the guanylate cyclase signaling cascade, activating protein kinase G leading to HSL phosphorylation and activation (Fig. 7.4). In addition to catecholamines, ANP directs fatty acid mobilization in response to aerobic conditioning (20). Although hormonal control of lipolysis via cAMP and cGMP signal transduction pathways are important contributors to plasma levels of FFA, TG appear to be more influenced by regulatory control of LPL, hepatic lipase (HL), and apolipoproteins such as apoA-IV, apoA-V, apoC-II, apoC-III, and apoE.
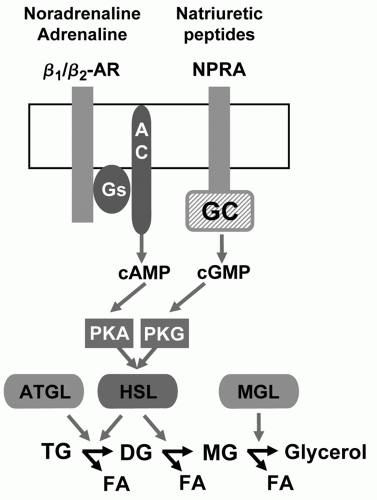 FIGURE 7.4 Model for lipolytic pathways in human adipose tissue. AC, adenylyl cyclase; AR, adrenoceptor; ATGL, adipose triglyceride lipase; DG, diglyceride; FA, fatty acid; GC, guanylate cyclase; Gs, stimulatory GTP-binding protein; HSL, hormone-sensitive lipase; MG, monoglyceride; MGL, monoglyceride lipase; NPRA, natriuretic peptide receptor A; PKA, protein kinase A; PKG, protein kinase G; TG, triglycerides. (Reproduced from Langin D. Adipose tissue lipolysis as a metabolic pathway to define pharmacological strategies against obesity and the metabolic syndrome. Pharmacol Res. 2006;53: 482-491, with permission). |
Insulin Resistance and Increased FFA
In models of insulin resistance, resulting from high fructose consumption or excess dietary fat, the increase in FFA stimulates de novo synthesis of TG in intestinal enterocytes, upregulation of MTP mass and activity, and enhanced chylomicron assembly and output (21). With saturation of adipose tissue stores and attenuation of insulin-mediated adipose tissue lipolysis, levels of plasma FFA are increased, which, in turn, are diverted to other tissues including liver, myocardium, and skeletal muscle. Increased plasma FFA levels may lead to sympathetic activation and increased blood pressure, oxidative stress, and endothelial dysfunction. FFA flux to the liver results in enhanced hepatic output of VLDL and gluconeogenesis, resulting in hypertriglyceridemia and potentially nonalcoholic steatohepatitis (NASH) (see following text) and/or pancreatic β-cell failure. In the myocardium, increased FFA may lead to myocardial steatosis or lipotoxicity with evidence of cardiac apoptosis, reduced ventricular contractility, and congestive heart failure (22), as well as susceptibility to ventricular arrhythmias. Likewise, in skeletal muscle, intramyocellular lipid accumulation has been observed in association with the metabolic syndrome and diabetes mellitus.
TG Levels in the Population
Based upon 3,982 fasting samples from the National Health and Nutrition Examination Survey conducted between 1999 and 2002, mean TG were 132 mg/dL (men) and 115 mg/dL (women), with an overall average of 123 mg/dL in U.S. adults aged 20 and older. However, while significant reductions have been observed in mean LDL-C (138 to 123 mg/dL between the initial survey years of 1976 to 1980), TG have increased by an
average of 8 mg/dL during this period. If the current trend continues, the majority of adult American men and women will eclipse the desirable level of fasting TG, defined as <150 mg/dL by the National Cholesterol Education Program (NCEP).
average of 8 mg/dL during this period. If the current trend continues, the majority of adult American men and women will eclipse the desirable level of fasting TG, defined as <150 mg/dL by the National Cholesterol Education Program (NCEP).
Pathophysiology of Hypertriglyceridemia
Elevation in TG is most commonly due to the combined effect of genetic predisposition with overriding metabolic or environmental perturbations. Much less commonly observed are monogenic disorders that result in chylomicronemia (see following text).
Familial Combined Hyperlipidemia (FCHL). Among the most common primary causes of elevated TG is FCHL, sometimes referred to as mixed hyperlipidemia. FCHL was initially described by Joseph Goldstein and colleagues from Seattle as a monogenic dyslipidemia in families of survivors of myocardial infarction (MI) (23). The disorder was completely expressed in adulthood, with delayed expression in children. FCHL has a pleiotropic presentation and can be expressed as one of three lipoprotein phenotypes (Fredrickson-Levy classification): elevated LDL-C and TG (type IIb phenotype), elevated LDL-C and normal TG (type IIa phenotype), or normal LDL-C and high TG (type IV phenotype) (see also Chapter 8). These phenotypes can be expressed at different times in the same patient, depending on factors such as weight gain or loss, control of diabetes, and kind of drug treatment (see also Chapter 8). When elevated TG are expressed in FCHL, low HDL-C and increased small, dense LDL particles are usually present. Diabetes and insulin resistance are often present in FCHL. The predominant mechanism is overproduction of apoB-containing lipoproteins (VLDL-IDL-LDL) and decreased clearance of remnant lipoproteins (chylomicron and VLDL remnants). No molecular defects have been proven as a cause of FCHL. Several candidate genes or loci have been postulated to play a role in FCHL, such as LPL, the APOA1/C3/A4/A5 gene cluster (see following text). Thus, FCHL is probably an oligogenic trait (see also Chapter 8).
Hyperapobetalipoproteinemia (hyperapoB). HyperapoB is a disorder that was first described by Sniderman and Kwiterovich in patients with angiographically defined coronary artery disease (CAD) who had normal to moderately elevated LDL-C (generally 120 to 160 mg/dL) but elevated apoB levels (see also Chapter 8). The TG can be normal (normotriglyceridemic hyperapoB) or elevated (hypertriglyceridemic hyperapoB) (24). The hyperapoB phenotype is due to an increased number of small, dense LDL particles that result from overproduction of VLDL. Recent studies indicate that hyperapoB, particularly hypertriglyceridemic hyperapoB, is the most common phenotype in patients with premature CVD. The primary defect in at least one group of hyperapoB patients is believed to be an intrinsic abnormality in a cell surface receptor for the acylation stimulatory protein, resulting in impaired FFA incorporation into TG in adipocytes (24). Consequently, increased flux of FFA to the liver augments hepatic VLDL production. HyperapoB patients often have increased FFA levels in the circulation, which may promote insulin resistance (see preceding text) and serve as a trigger to arrhythmias and sudden cardiac death.
There are no pathognomonic clinical signs associated with FCHL or hyperapoB. Tendon xanthomas are not present; however, isolated xanthelasmas can be found. Corneal arcus and vertical creases in the ear lobes have been described in these disorders.
Familial Hypertriglyceridemia (FHT). FHT usually presents with elevated TG and normal LDL-C (type IV lipoprotein phenotype). The disorder is expressed completely as an autosomal dominant in affected adult members from kindreds with hypertriglyceridemia. Unlike patients with FCHL or hyperapoB, elevated VLDL in subjects with FHT is not due to VLDL overproduction but due to delayed catabolism of TG-rich lipoproteins, often due to downregulation of LPL. LDL-C and apoB levels are therefore not elevated, and risk of premature CVD is lower in FHT than in FCHL and hyperapoB. TG in untreated subjects can vary from 200 mg/dL to >2,000 mg/dL, and pancreatitis can occur.
Familial Dysbetalipoproteinemia (FDBL). FDBL is a relatively uncommon disorder that is expressed by elevated remnant lipoproteins. VLDL in FDBL is enriched in cholesterol compared to TG, giving an elevated ratio of VLDL cholesterol to TG (>0.30; average normal, 0.2). VLDL are isolated by ultracentrifugation at d 1.006 g/ml and subjected to electrophoresis. VLDL from a patient with FDBL have a β- rather than a pre-B migration on electrophoresis (termed “floating β-lipoproteins”) (see also Chapter 2). The combination of an elevated VLDL cholesterol to TG ratio and the presence of floating β lipoproteins has been referred to as a type III lipoprotein phenotype in the Fredrickson-Levy classification system. The most common form of FDBL is due to a molecular variant in the apoE gene (Arg145 → Cys) in the homozygous state (apoE2/E2). This is a necessary but insufficient condition for FDBL (apoE2/E2 is present in about 1/100 in the general population). Overproduction of VLDL must also be present. ApoE2/E2 on the TG-rich lipoproteins constitutes a deficient ligand for their binding to LRP and LDL-R on the surface of the liver, leading to the reduced hepatic uptake of particles of both exogenous and endogenous origin. Patients with FDBL are therefore very sensitive to dietary fat and weight. Metabolic triggers for FDBL include hypothyroidism and diabetes mellitus. In addition to the common form of FDBL, there is a rare form inherited as a dominant disorder in which there are mutations in the apoE gene other than the E2 variant. The expression of FDBL in this group is similar to those with the common “recessive” form, except that it is also expressed in children (see also Chapter 12). The biochemical diagnosis of FDBL can be difficult (see also Chapter 2).
Patients with FDBL have marked increases in both total cholesterol (TC) and TG, often >300 mg/dL, and can be mistaken for those with FCHL. Unlike patients with FCHL and hyperapoB, however, those with FDBL have normal apoB levels. The clinical presentation can provide a clue to diagnosis. Patients with FDBL often present with striking planar xanthomas in the creases of the palms and tuberoeruptive xanthomas in the elbows and buttocks. Tendon xanthomas are less common but have also been reported. The xanthomas can disappear dramatically on combined treatment of a low-fat diet, weight reduction, and use of fibric acid derivatives. Fibrates can help distinguish between FCHL and FDBL; patients with FCHL manifest a marked lowering of TG but an increase in TC and LDL-C, while those with FDBL normalize both their TG and TC. Niacin and statins are also effective. Postmenopausal women respond dramatically to estrogen.
TABLE 7.1 CAUSES OF HYPERTRIGLYCERIDEMIA | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Related posts:
 Pathophysiology and Vascular Biology of Atherosclerosis
Dyslipidemia in Children and Adolescents
Fish Oils in the Treatment of Dyslipidemia and Cardiovascular Disease
Use of Noninvasive Methods to Diagnose Cardiovascular Disease in Dyslipidemic Patients
Clinical Utility of Inflammatory Markers
Pharmacologic Treatment of Dyslipidemia and Cardiovascular Disease
Pathophysiology and Vascular Biology of Atherosclerosis
Dyslipidemia in Children and Adolescents
Fish Oils in the Treatment of Dyslipidemia and Cardiovascular Disease
Use of Noninvasive Methods to Diagnose Cardiovascular Disease in Dyslipidemic Patients
Clinical Utility of Inflammatory Markers
Pharmacologic Treatment of Dyslipidemia and Cardiovascular Disease
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


