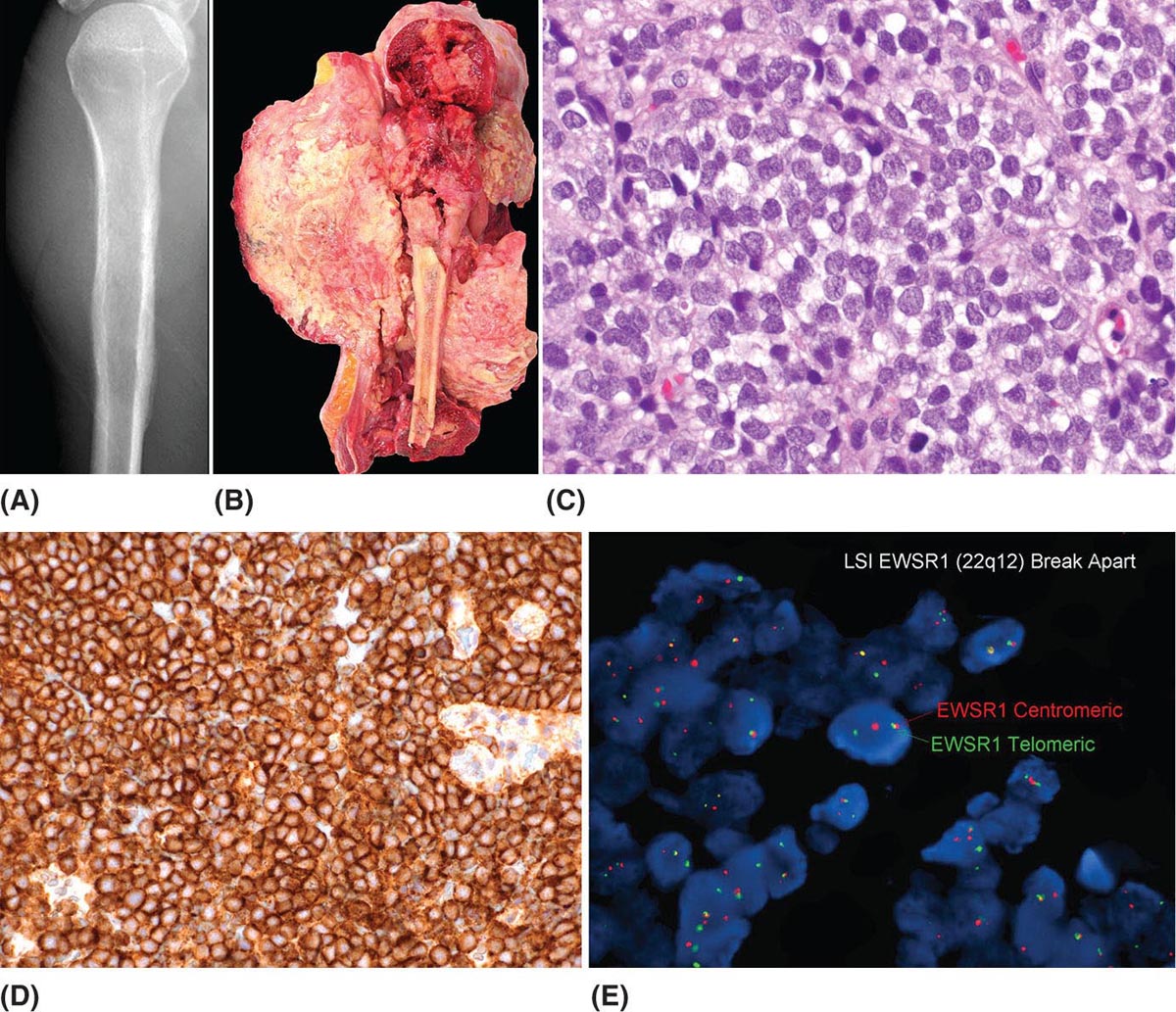
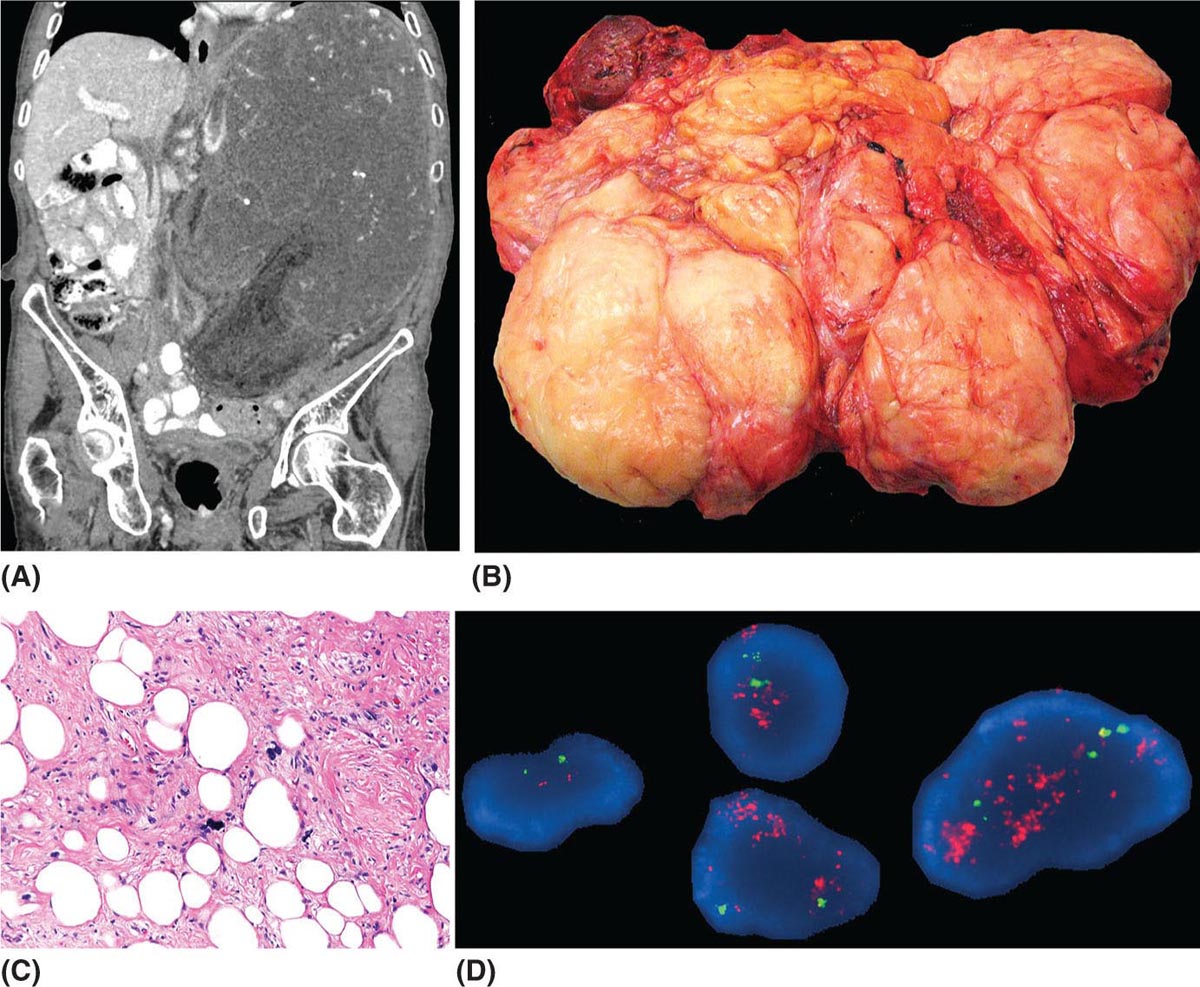
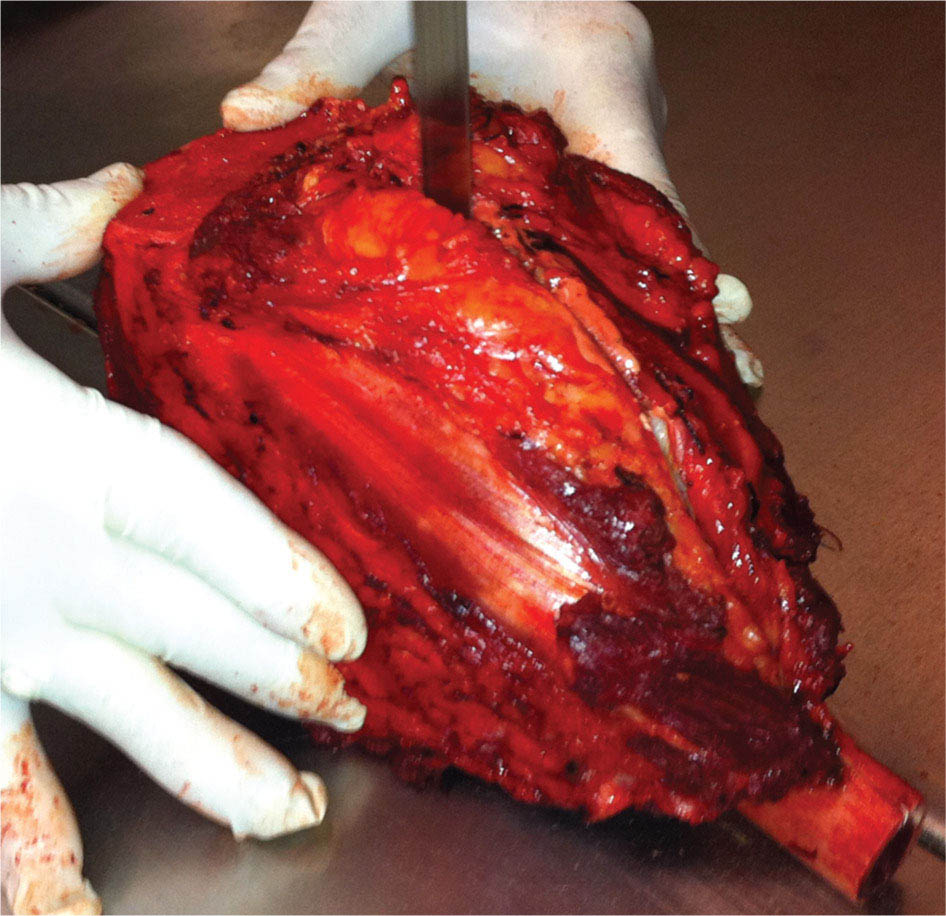
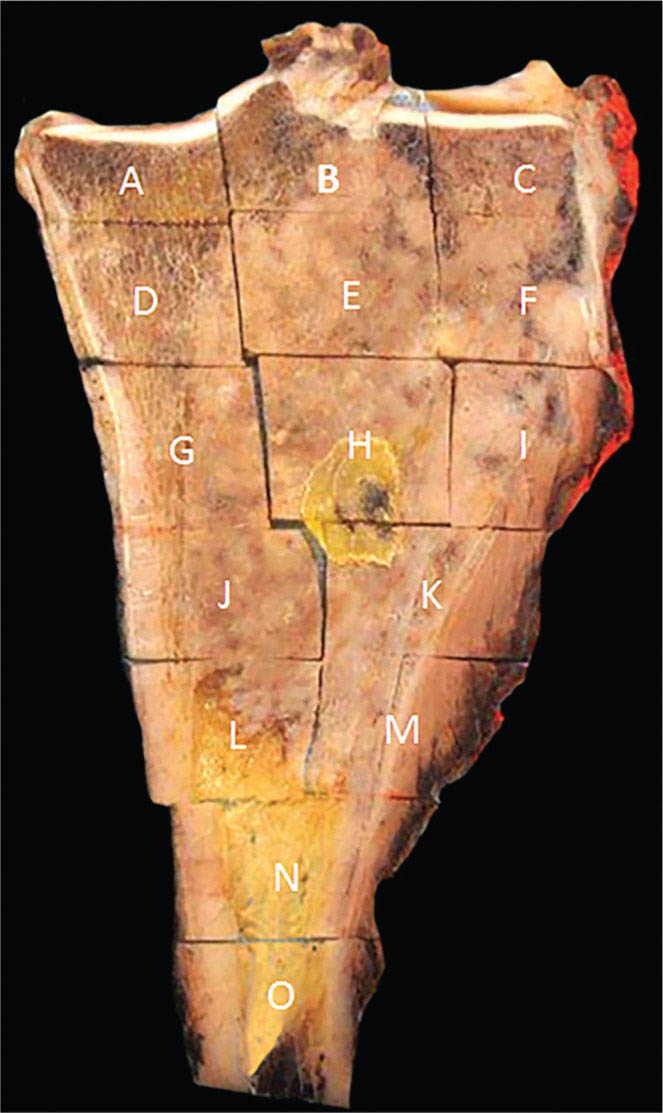
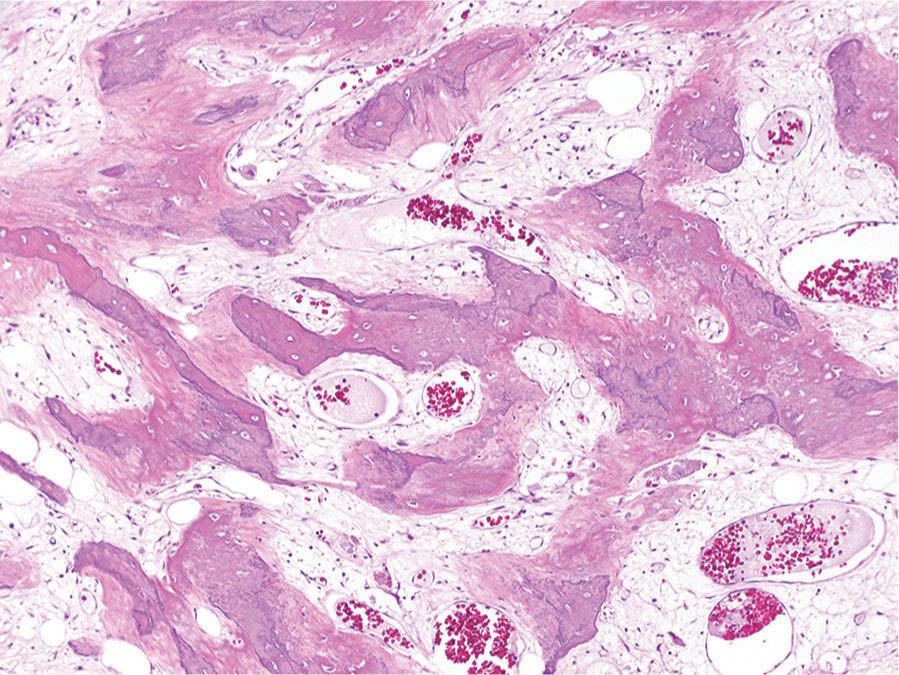
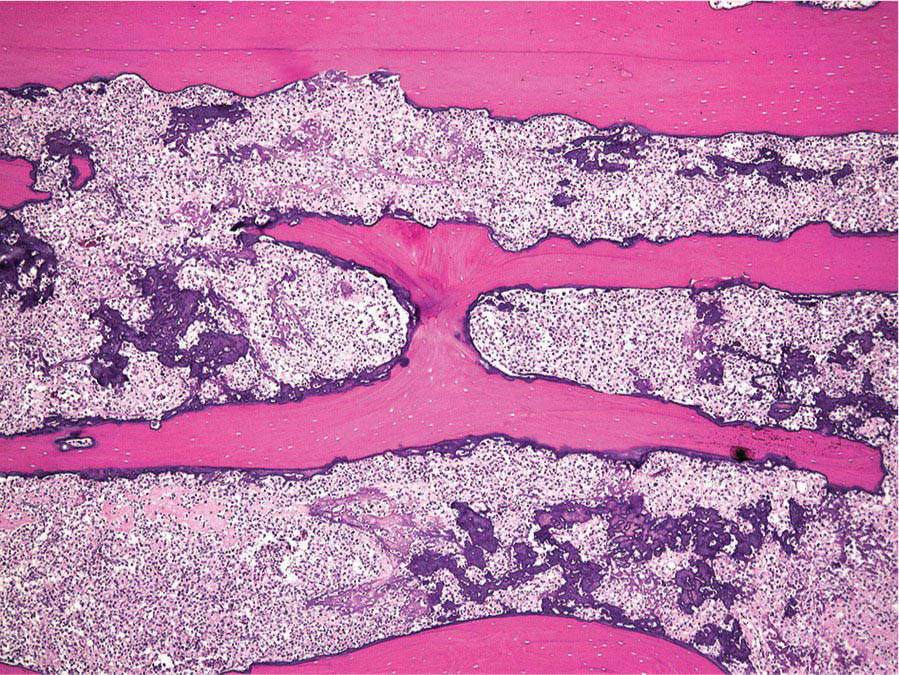
273 Pathology of Sarcoma Sarcoma is defined as a malignant neoplasm whose phenotype recapitulates cells of mesodermal derivatives including the connective tissue, muscle, and peripheral nerve sheath. Biologically, sarcomas have the potential to progressively enlarge, grow with an infiltrative pattern, and metastasize. This chapter discusses the classification, clinical manifestations, diagnosis, and prognosis of bone and soft tissue sarcoma. The clinical and morphologic diversity observed in sarcoma is challenging. The diagnosis, classification, prognostication, identification of important biomarkers, and staging of these neoplasms require the integration of radiologic, morphologic, immunohistochemical, and molecular analyses. The identification of genetic abnormalities, biomarkers, susceptibility to immune therapy, and assessment of treatment effect are becoming increasingly important in the management of these neoplasms. The experience and skills required to optimally manage patients with sarcoma reside in the dedicated sarcoma multidisciplinary team. Sarcoma, Connective and Soft Tissue Neoplasms, Pathology, Classification, Genetics biomarkers, diagnosis, immunohistochemical analyses, molecular analyses, morphologic analyses, neoplasms management, prognostication, radiologic analyses, sarcoma Biomarkers, Diagnosis, Disease Management, Immunohistochemistry, Morphological and Microscopic Findings, Radiology, Sarcoma INTRODUCTION The word sarcoma is derived from the Greek term “sarkoun,” which means “fleshy substance or excrescence.”1 Sarcoma is defined as a malignant neoplasm whose phenotype recapitulates cells of mesodermal derivatives including the connective tissue, muscle, and peripheral nerve sheath. Sarcoma arises in any portion of the body; however, most originate in the soft tissues and skeleton.2 Biologically, sarcomas have the potential to progressively enlarge, grow with an infiltrative pattern, and metastasize.3 Most sarcomas disseminate via the vascular system, with the lungs being the most common site for metastatic disease.4 Lymph node metastases from sarcomas are uncommon and occur in approximately 3.5% of cases.5 Tumors associated with the greatest risk of spread to lymph nodes include epithelioid sarcoma and clear cell sarcoma followed by angiosarcoma and synovial sarcoma.6 The defining biologic behavior of sarcoma dictates treatment, which includes surgery, radiation, and systemic therapy. CLASSIFICATION The current 2013 World Health Organization (WHO) classification recognizes more than 100 soft tissue and bone tumor entities, and more than 70 are sarcomas.7 The features used to classify sarcomas are their normal tissue counterpart, and this group accounts for the vast majority (Table 3.1). A comparatively smaller number of sarcomas have consistent and distinctive clinicopathologic features, but lack an identified normal tissue counterpart, and their classification is based on their morphologic features (spindle cell sarcoma, small round cell sarcoma)7,8 and their molecular aberrations (EWSR1/FUS–NFATc2-rearranged round cell sarcoma).9 EPIDEMIOLOGY Sarcomas are relatively infrequent compared to carcinomas.10 In 2018, sarcomas represented approximately 1% of all cancers in the United States, with approximately 13,000 to 16,000 new cases and an overall incidence of 7.1 cases per 100,000 people. They are among the top five causes of cancer deaths for those under 20 years of age, and approximately 5,000 to 6,000 patients die of their sarcomas annually in the United States.11 Most cases are high grade (33.6%) and >5 cm (51.3%), and the 5-year survival rate is estimated to be 71.4%.11,12 CLINICAL MANIFESTATIONS Clinical features associated with sarcoma depend on their location, size, and rate of growth. Common findings are a progressively growing, deep-seated, firm, and fixed mass that is often associated with pain of varying degrees of severity (Figures 3.1 and 3.2). Some tumors may be superficial, mobile, and asymptomatic. Sarcomas arising in the retroperitoneum, abdomen, or pelvis may achieve enormous size before they produce symptoms (Figure 3.3). Tumors developing in bone may present with a devastating pathologic fracture.13 Occasionally, sarcomas are associated with symptoms secondary to the effects of their metabolic products that cause fever, anemia, lethargy, weight loss, and histamine-like reactions.14 28TABLE 3.1 Major Groups of Sarcoma PEComa, perivascular epithelioid cell tumor. DIAGNOSIS Preoperative Biopsy Primary diagnosis usually requires tissue biopsy to obtain samples of the tumor for histologic, immunohistochemical, and molecular analysis.15,16 The biopsy site should be selected such that subsequent management of the tumor is not affected. A minimally invasive technique to obtain diagnostic tissue is needle biopsy and this procedure may need to be performed under radiologic guidance—areas of suspected tumor necrosis should be avoided. A minimum of three tumor-bearing cores of tissue is usually sufficient, and there should be consideration for performing frozen section analysis on a portion of one core while the patient is still available to confirm that the diagnostic tissue is retrieved; this can also expedite tissue testing (touch preps for fluorescent in situ hybridization [FISH]) and patient triage.16,17 Fine-needle aspiration cytologic biopsy is usually reserved for documenting recurrence or metastasis. It should be kept in mind that sarcomas are notoriously heterogeneous, and there is a significant potential for sampling error. A minority of tumors may require open biopsy because of failed attempts at needle biopsy, and small superficial tumors may be adequately removed with excisional biopsy by an experienced sarcoma surgeon.17,18 FIGURE 3.1 Large osteosarcoma of proximal humerus presenting as painful mass. FIGURE 3.2 (A) Radiograph of Ewing sarcoma showing poorly defined mass in proximal diaphysis of humerus. (B) Resected tumor is pink, tan, and fleshy, and destroys the bone and extends into the soft tissues. (C) Tumor is composed of sheets of small round cells with little cytoplasm. (D) Immunohistochemical stain for CD99 shows that tumor cells exhibit strong membranous staining. (E) FISH using EWSR1 break-apart probe shows separation of signals indicative of rearrangement. FISH, fluorescence in situ hybridization. FIGURE 3.3 (A) Reformatted coronal CT image of large well-differentiated liposarcoma filling the left side of retroperitoneum and pelvis. (B) Resected tumor composed of lobule of soft yellow fat. (C) Neoplastic cells are adipocytes with enlarged hyperchromatic nuclei. (D) FISH for amplification of MDM2 shows numerous copies of the gene. FISH, fluorescence in situ hybridization. Resection Specimens Surgically excised specimens should be sent to the pathology laboratory in a fresh state directly after removal from the patient. Ideally, the surgeon and pathologist review the specimen together to clarify orientation and proximity of tumor to important anatomic structure, and to identify and assess surgical margins. This can be performed in real time, so that preliminary margin status can be determined and the surgical team can decide whether or not additional tissue removal is necessary to achieve a negative margin. The resected tumor should be cut along its longest axis (Figure 3.4), photographed, and the margins serially sectioned in a perpendicular plane. A small sample of tumor and normal tissue can be collected for biobanking.19 The specimen should then be thinly bread-loafed at 0.5 to 1 cm intervals; described, indicating color, consistency, presence of cysts, hemorrhage, and necrosis; and placed in formalin in an adequately sized container and allowed to fix for at least 12 hours. The guideline for tumor sampling is one block of tissue per centimeter of greatest tumor dimension.20 Bone tumors should be decalcified after fixation, and a portion of viable tumor should be decalcified in an agent that does not damage RNA or DNA, such as EDTA.21 Resected tumors that have been treated with preoperative chemotherapy (osteosarcoma, Ewing sarcoma, dedifferentiated chondrosarcoma, mesenchymal chondrosarcoma, high-grade fibrosarcoma, and high-grade soft tissue sarcomas) require determination of percent tumor necrosis. To accomplish this, the central slab of tumor can be mapped and blocked out in its entirety (Figure 3.5). A section of tumor per centimeter (as determined by its greatest dimension) should be processed from each of the remaining two hemispheres of the specimen. During histologic review, the amount of tumor necrosis on each slide can be estimated and these scores can then be averaged to calculate the overall percentage of tumor necrosis (Figure 3.6). The areas of viable and necrotic tumor can then be located on the map of the slab section, if necessary. FIGURE 3.4 Osteosarcoma being cut through its long axis parallel to the underlying bone. FIGURE 3.5 Central slab of resected osteosarcoma mapped prior to processing. FIGURE 3.6 Osteosarcoma treated with neoadjuvant therapy producing 100% necrosis. The residual trabeculae of neoplastic bone are devoid of tumor cells. Microscopic Examination Accurate diagnosis depends on having an adequate quantity of viable tumor to assess architecture, relationship to nonneoplastic tissue, cytologic features, and mitotic activity including atypical forms and necrosis.22 Criteria of malignancy commonly include hypercellularity, nuclear pleomorphism and hyperchromasia, abundant mitotic activity with structurally atypical forms, necrosis, vascular invasion, and the presence of metastasis.22,23 An additional feature of bone tumors is the presence of an infiltrative growth pattern within the medullary cavity with encasement of preexisting bone trabeculae (Figure 3.7). An experienced pathologist can differentiate benign from malignant tumors with a sensitivity of 99.4% and a specificity of 98.7%.24 Once a differential diagnosis is made, immunohistochemistry, cytogenetics, and molecular studies can be performed to establish a precise diagnosis.14,23,25 Grading of Bone and Soft Tissue Sarcomas The pathologist’s attempt to predict the biologic behavior of sarcoma is reflected in the histologic grade and this is translated into the biologic grades of low and high.15 In other words, grading assesses the aggressiveness of the sarcoma and its risk of metastasis. Histologic grading is the single most significant prognostic factor of sarcoma and, as such, is a major independent predictor of metastasis26 (Table 3.2). Histologic typing alone provides sufficient information for predicting the clinical course in select sarcomas such as synovial sarcoma, Ewing sarcoma, mesenchymal chondrosarcoma, low-grade fibromyxoid sarcoma, well-differentiated liposarcoma, and so on; however, many require formal histologic grading. FIGURE 3.7 Osteosarcoma infiltrating the marrow cavity and surrounding individual bony trabeculae.
Phenotype
Type
Adipocytic
Atypical lipomatous tumor/well-differentiated liposarcoma
Dedifferentiated liposarcoma
Myxoid liposarcoma
Pleomorphic liposarcomas
Fibroblastic/myofibroblastic
Low-grade myofibroblastic sarcoma
Myxoinflammatory fibroblastic sarcoma
Infantile fibrosarcoma
Adult fibrosarcoma
Infantile fibrosarcoma
Myxofibrosarcoma
Low-grade fibromyxoid sarcoma
Sclerosing epithelioid fibrosarcoma
So-called fibrohistiocytic tumors
Malignant giant cell tumor of soft tissue
Smooth muscle/pericytic
Leiomyosarcoma
Malignant PEComa
Skeletal muscle
Embryonal rhabdomyosarcoma
Alveolar rhabdomyosarcoma
Pleomorphic rhabdomyosarcoma
Spindle cell/sclerosing rhabdomyosarcoma
Vascular
Retiform hemangioendothelioma
Papillary intralymphatic angioendothelioma
Composite hemangioendothelioma
Kaposi sarcoma
Pseudomyogenic hemangioendothelioma
Epithelioid hemangioendothelioma
Angiosarcoma
Bone forming
Conventional osteosarcoma
Well-differentiated intramedullary osteosarcoma
Parosteal osteosarcoma
Periosteal osteosarcoma
High-grade surface osteosarcoma
Secondary osteosarcoma
Extraskeletal osteosarcoma
Cartilage forming
Conventional chondrosarcoma
Dedifferentiated chondrosarcoma
Clear cell chondrosarcoma
Mesenchymal chondrosarcoma
Gastrointestinal stromal tumor
Malignant gastrointestinal stromal tumor
Nerve sheath
Malignant peripheral nerve sheath tumor
Malignant granular cell tumor
Ectomesenchymoma
Small round cell sarcoma
Ewing sarcoma and related tumors
Melanotic neuroectodermal tumor
Notochordal tumors
Chordoma
Undifferentiated sarcoma
Round cell sarcoma
Pleomorphic spindle cell sarcoma
Epithelioid cell sarcoma

Related posts:



Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


