Pathogenesis of Graves’ Disease
Terry F. Davies
Introduction
Robert Graves’ disease is a uniquely human autoimmune disease with stimulating autoantibodies to the thyrotropin (TSH) receptor as the major pathogenic feature. Unlike many autoimmune diseases, these antibodies are not just markers of the disease but are responsible for the hyperthyroidism that occurs in most patients. Both B- and T-lymphocytes are known to be directed at three well-characterized thyroid autoantigens: Thyroglobulin (Tg), thyroid peroxidase (TPO), and the TSH receptor (TSHR). However, much evidence suggests that it is only the TSHR which is the primary autoantigen of Graves’ disease and that immune responses to other thyroid antigens are only reflective of a concomitant thyroiditis.
Autoimmunity
Definition of an Autoantigen
There are a number of simple rules concerning the self-molecules (autoantigens) with which T cells and autoantibodies interact. Autoantigens are present from birth and do not just appear during later development. In fact, autoantigens are highly conserved structural proteins coded for by genes with low mutation rates. Hence, autoantigens are not abnormal molecules but they may be coded for by polymorphic genes within the population. Such polymorphisms may cause structural and functional variations that may be important for the clinical phenotype (1). However, autoimmune diseases are thought to primarily involve defects in immune surveillance pathways which normally maintain tolerance.
The Nature of T Cells
Self-reactive T cells are deleted within the thymus gland where all autoantigens, including thyroid autoantigens such as the TSH receptor, are expressed (2,3) under the control of the AIRE gene (4). The T cells that survive both intrathymic deletion and peripheral deletion are a complex mixture of cells of different phenotypes and many such T cells are in transition between immature and mature forms. T cells exert their function by the secretion of cytokines and studies of cytokine secretion have provided useful criteria to help understand the way T cells initiate and control the immune response. The CD4+ T cells have been shown to be of four principal types (T helper cell type 1, T helper cell type 2, T regulatory cells, and Th17 cells). These T-cell subsets differ functionally in their pattern of cytokine secretion and differ phenotypically in their pattern of chemokine receptor expression (Table 18A.1). CD8+ T cells tend to be primarily cytotoxic T cells able to kill target cells but also have regulatory capacity.
Table 18A.1 Subsets of CD4+ T Cells | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
Antibody and T-cell Interactions with Antigen
Antibodies are immunoglobulins which are present in the serum or expressed on the surface of B cells and are able to bind to their specific antigenic molecules directly. The strength of this binding is measured as the affinity of the antibody and is directly dependent, to a large degree, on the number of antigen binding sites (known as epitopes). Hence, the binding energy is usually greater for conformational antigens with multiple binding sites than for the binding to a small linear peptide region on an antigen. Pathogenic antibodies of high affinity are, therefore, most likely to interact with conformational antigens and this is an important concept in understanding Graves’ disease where stimulating TSHR antibodies only interact appropriately with the correctly folded ectodomain of TSH receptors. The T-cell antigen receptor is also a member of the immunoglobulin family, but it has a transmembrane domain that anchors it within the cell surface. The CD8+ T cells recognize antigenic peptides complexed with Class I HLA molecules (A, C, B) and CD4+ T cells recognize antigenic peptides complexed with Class II HLA molecules (DR, DP, DQ) (Fig. 18A.1). The T cells recognize small linear peptides within the antigen–HLA complex and, therefore, are termed “HLA restricted.” In thyroid autoimmunity, thyroid antigens are first engulfed by antigen-presenting cells (APCs) such as macrophages and dendritic cells and then digested by proteases within these cells. Antigen breakdown products (peptides) are then bound to intracellular HLA molecules and the complexes are transported onto the cell surface by transporters associated with antigen processing
(TAPs). There is evidence that thyroid cells themselves may act as APCs (see later).
(TAPs). There is evidence that thyroid cells themselves may act as APCs (see later).
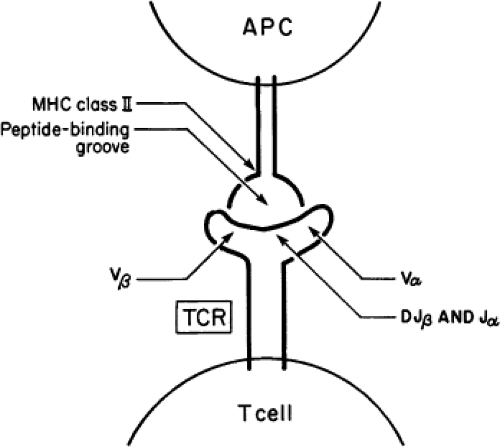 Figure 18A.1 Diagram of the mechanism of antigen presentation by an APC to the T-cell antigen receptor (TCR) of a T cell. |
Second Signals
Both B cells and T cells rely on secondary signals once the antigen has been identified, to enter a proliferative and secretory state (5). A variety of cytokines secreted by T cells and APCs serve as second B-cell signals. In contrast, T cell second signals are supplied by signal transduction from cell-surface molecules (such as the B7 family and CD40) (Table 18A.2) that are activated by ligands on the surface of the APCs (CD28 and CD40L, respectively). B cells and T cells that interact with specific antigens in the absence of a second signal become desensitized, a state referred to as anergy. Hence, anergy is one of the mechanisms that may result in suppression of an immune response.
Table 18A.2 Second Signals for T Cell Activation | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
Criteria for an Autoimmune Disease
Three types of evidence can be marshaled to establish that a human disease is autoimmune in origin:
Direct evidence is provided if the disease, or manifestations of the disease, arise from transfer of pathogenic antibody or pathogenic T cells to humans or animals.
Indirect evidence may be based on studies in experimental animals by immunization with candidate antigens and a disorder similar to the human disease ensues.
Circumstantial evidence may be obtained from clinical studies demonstrating, e.g., an immune infiltrate at the site of disease or the presence of an associated autoimmune disease.
In Graves’ disease the antibody and T-cell–mediated thyroid antigen–specific immune responses are well defined and there are now induced animal models of this uniquely human disease that reproduce the hyperthyroidism of the disease. Historically, the induction of thyroid hyperfunction by TSHR antibodies in normal subjects following the transfer of serum from patients with Graves’ disease (6) was the first direct proof for an autoimmune origin of a human disease. Another early demonstration of the in vivo effects of TSHR antibodies came from studies demonstrating stimulation of the thyroid in utero by transplacental passage of TSHR antibodies (7).
Restriction versus Polyclonality
It is now possible to learn whether autoimmune reactions are multireactive and more likely to be representative of a secondary polyclonal immune response, or whether the immune response is much more focused, involving restricted sets of B cells and T cells (8). In an autoimmune disease, the immune system is where the abnormality is to be found and, therefore, the primary autoimmune response at the onset of the disease should be oligoclonal. This has, indeed, proven to be the case for most of the human autoimmune diseases, including Graves’ disease, as discussed below.
Thyroid Immunology
Intrathyroidal B Cells
The B cells that accumulate within the thyroid gland of patients with Graves’ disease have reduced proliferative responses to B-cell mitogens and greater basal immunoglobulin secretion than peripheral blood B cells indicative of their activated state. B cells from Graves’ thyroid tissue secrete thyroid autoantibodies spontaneously in vitro, again implying pre-activation. Indeed, the thyroid gland is the primary site of thyroid autoantibody secretion in autoimmune thyroid disease (AITD) (Fig. 18A.2), as evidenced by the decline in thyroid autoantibodies after antithyroid drug treatment or thyroidectomy (9,10).
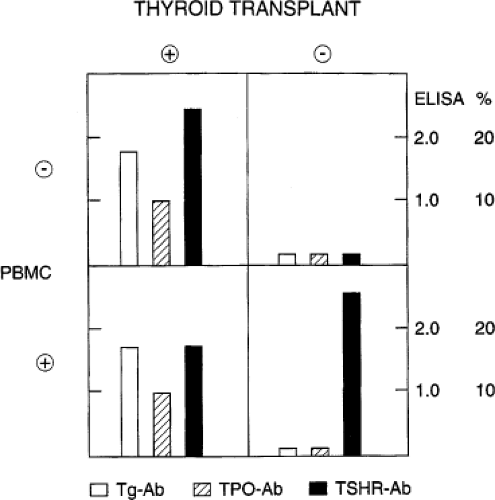 Figure 18A.2 Production of antibodies in mice with subacute combined immunodeficiency disease (SCID) after thyroid tissue engraftment or intraperitoneal injection of peripheral blood mononuclear cells (PBMC) from a patient undergoing surgery for the treatment of Graves’ disease. Thyroid antibodies were measured in mouse serum 4 to 6 weeks after transplantation. In the absence of thyroid tissue and PBMC, no thyroid antibodies were detectable (upper right). In contrast, transplantation of thyroid tissue resulted in the production of Tg, TPO, and TSH receptor (TSHR) antibodies (Ab) by the mice. |
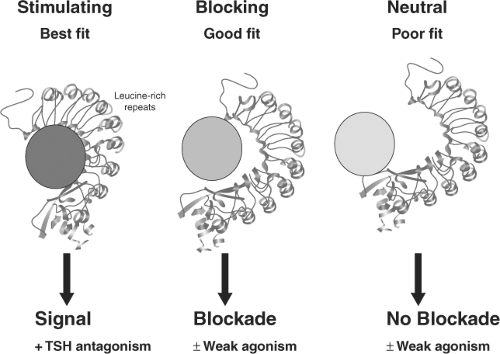 Figure 18A.3 The three varieties of TSH receptor antibodies. |
Autoantibodies to the Human TSH Receptor
The long-acting thyroid stimulator (LATS) was discovered by Adams and Purves almost 50 years ago during a search for thyroid-stimulating activity in the serum of patients with Graves’ disease using a bioassay for pituitary TSH (11). The patient’s serum stimulated radioiodine release from pre-labeled thyroid glands of guinea pigs for a much longer time period than did a pituitary TSH preparation (hence the term long-acting). This prolonged stimulating activity was then found to reside in the IgG fraction of serum. With the advent of biologically active radiolabeled TSH, it became possible to detect TSH receptors on thyroid membranes, and subsequently this
IgG activity in Graves’ patients was found to compete with TSH for receptor occupancy proving that it contained TSHR antibodies acting as TSH agonists (12). Hence, in patients with Graves’ disease, the thyroid gland is no longer under the control of pituitary TSH but is continuously stimulated by circulating antibodies with TSH-like activity. It is not possible, therefore, to suppress the thyroid gland using exogenous thyroxine suppression of any remaining endogenous TSH because the thyroid-stimulating antibodies are unaffected.
IgG activity in Graves’ patients was found to compete with TSH for receptor occupancy proving that it contained TSHR antibodies acting as TSH agonists (12). Hence, in patients with Graves’ disease, the thyroid gland is no longer under the control of pituitary TSH but is continuously stimulated by circulating antibodies with TSH-like activity. It is not possible, therefore, to suppress the thyroid gland using exogenous thyroxine suppression of any remaining endogenous TSH because the thyroid-stimulating antibodies are unaffected.
Bioactivity of TSHR Antibodies
It is now known that antibodies that bind to the TSH receptor may or may not initiate an intracellular signal. Those antibodies which induce a strong signal are referred to as TSHR-stimulating and those which induce weak or no signals are TSHR-blocking or TSHR-neutral antibodies (Fig. 18A.3) (13). Further complicating this issue has been the observation of the simultaneous presence of TSHR-stimulating and TSHR-blocking antibodies in the same serum samples from patients with Graves’ disease. The effective degree of thyroid stimulation under such circumstances is dependent on the relative concentration and bioactivities of the different antibodies.
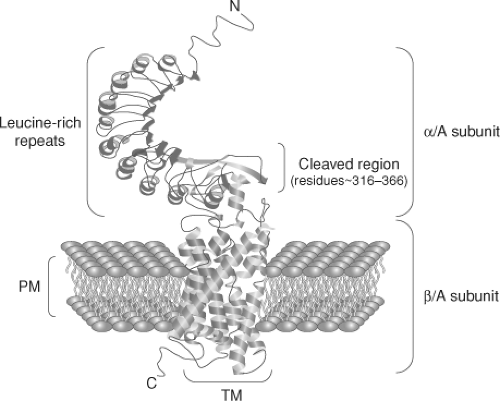 Figure 18A.4 A model of the TSH receptor. Note the α and β subunits of the receptor which are disulfide linked and formed by protease cleavage of the intact molecule. The α subunit is subsequently thought to be shed into the circulation where it most likely contributes to the maintenance of tolerance to the TSHR. |
Prevalence of TSHR Antibodies in Graves’ Disease
TSHR antibodies are detectable only in patients with AITD. Such antibodies are, therefore, disease specific, in great contrast to the high prevalence of Tg and TPO antibodies that may occur in people who manage to retain apparently normal thyroid function. Eighty to 100% of untreated patients with thyrotoxicosis caused by Graves’ disease have detectable TSHR antibodies in their serum depending on the sensitivity of the assay used (14,15,16). The titers of TSHR antibodies may be reduced by treatment of the patient’s thyrotoxicosis and when they persist in high concentrations they often predict a recurrence
after withdrawal of antithyroid drug treatment unless thyroiditis or iodine deficiency prevent the thyroid gland from responding (17,18) (Table 18A.3). TSHR-blocking antibodies may, in time, become the more prevalent antibody after treatment of thyrotoxic patients with Graves’ disease, contributing to the development of later thyroid failure (19).
after withdrawal of antithyroid drug treatment unless thyroiditis or iodine deficiency prevent the thyroid gland from responding (17,18) (Table 18A.3). TSHR-blocking antibodies may, in time, become the more prevalent antibody after treatment of thyrotoxic patients with Graves’ disease, contributing to the development of later thyroid failure (19).
Table 18A.3 Factors Influencing Recurrence of Graves’ Disease | ||||||
|---|---|---|---|---|---|---|
|
Immunologic Characteristics of TSHR Antibodies
TSHR antibodies are oligoclonal. Patients with Graves’ disease demonstrate TSHR-Abs with light chain restriction, and the TSH agonist bioactivity is found mostly in the IgG1 subclass (20). The cloning and subsequent crystallization of the TSHR has permitted the initiation of detailed studies of its epitopes and structure-function relationships (21,22,23,24,25). The large extracellular domain of the TSHR is the major immunogenic region, and stimulating TSHR antibodies bind primarily, but not exclusively, to the leucine-rich repeats in this domain of the receptor (see Fig. 18A.4). The difference in functional activity of different TSHR antibodies likely relates to their molecular binding characteristics inducing structural changes in receptor conformation. For example, bioactive and high affinity stimulating antibodies to the TSHR interact with highest affinity to the receptor ectodomain alone (26), while less active TSHR antibodies recognize linear epitopes in the N terminal and “hinge” region. Many of the latter antibodies are blocking or neutral in their competition with TSH binding and may be weak agonists (27).
TSH Receptor Function in Graves’ Disease
Similar to TSH, TSHR-stimulating antibodies activate the TSHR to induce binding of Gs and Gq proteins and activate both cAMP-mediated signal transduction and the IP3 cascade (28). The result is release and synthesis of thyroid hormones, the synthesis of Tg, and stimulation of iodine uptake, protein synthesis, and thyroid cell growth. However, chronic treatment of mice with high concentrations of stimulating TSHR-Ab did not produce severe hyperthyroidism as expected but a relatively mild degree of thyroid overactivity secondary to desensitization of the host thyroid (29). Hence, the low levels of TSHR-Ab in Graves’ patients may explain why the thyroid is able to continue to over secrete thyroid hormone for many years. Recent evidence suggests that some neutral TSHR-Abs may activate Gq binding only and, in the absence of cAMP activation, this leads to thyroid cell apoptosis under appropriate conditions (27).
Lessons from Monoclonal TSHR Antibodies with Stimulating Activity
For many years, the induction of animal models of Graves’ disease produced TSHR antibodies which only blocked the TSHR. This was before it was understood that the TSHR must be conformationally correct to induce stimulating antibodies (30). High affinity TSHR-stimulating monoclonal antibodies are now available following immunization with TSHR cDNA directly or within an adenovirus vector (31,32) (Fig. 18A.5). These mAbs have become new probes for the TSHR and have also allowed the crystallization of the TSHR ectodomain (24). As discussed earlier, stimulating antibodies only interact with the natural, fully conformed receptor and all appear to share many of the same conformational epitopes.
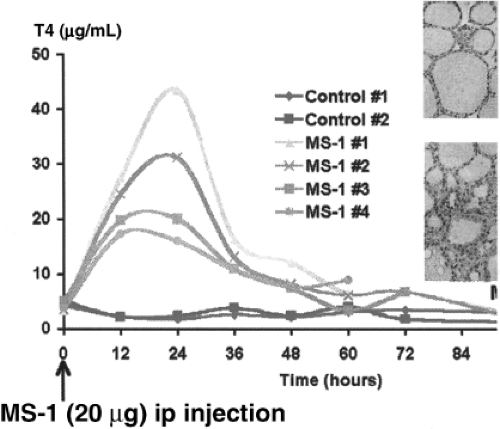 Figure 18A.5 In vivo thyroid stimulation by monoclonal thyroid-stimulating antibody. 20 μg of antibody (MS-1) was administered intraperitoneally and the serum thyroid hormone response measured at the illustrated time intervals. The insets show the histologic response of the thyroid gland with thyroid cell hypertrophy and colloid droplet formation. |
Other Thyroid Antibodies in Graves’ Disease
The majority of patients with Graves’ disease have circulating antibodies to additional thyroid autoantigens and not just the TSHR. In particularly, Tg and TPO antibodies, sometimes in high titers, are found in the majority of such patients. However, these antibodies are polyclonal in nature (33) and, therefore, are a secondary immune response and most likely reflect a coincidental, but controlled, autoimmune thyroiditis which may be seen histologically. Indeed, 20% to 30% of patients with Graves’ disease treated only with antithyroid drugs become hypothyroid long after antithyroid drug therapy (19). Hence, Graves’ disease appears to develop on a background of, or coincidental with, autoimmune thyroiditis.
The Intrathyroidal Lymphocytic Infiltrate
Much of the early evidence that Graves’ disease was an autoimmune disease is based on the discovery of TSHR antibodies. However, supporting evidence was available much earlier within the thyroid gland itself. The thyroid in Graves’ disease is characterized by a lymphocytic infiltration which is heterogeneous and suggestive of differing degrees of antigenicity between follicles. Evidence in support of this explanation is the finding of thyroid follicular hyperplasia which tends to be more extensive in areas of infiltration (34,35) (Fig. 18A.6) and areas where apoptotic thyroid cells can be found (36). Antithyroid drug treatment may markedly reduce the degree of lymphocytic infiltration, which should be kept in mind when examining individual patient samples. Although the intrathyroidal lymphocyte population is mixed, immunohistologic staining and functional studies have shown that the majority of cells are T cells and that B-cell germinal centers are much less common than in chronic autoimmune thyroiditis (Hashimoto’s disease) (37,38). Intra-epithelial T cells and plasma cells can be seen both adjacent to and within the thyroid follicles (in peripolesis) (37). There is none of the follicular destruction seen in Hashimoto’s disease despite the lymphocytic infiltration. In fact, as mentioned earlier, thyroid follicular epithelial cell size has been correlated with the intensity of the local infiltrate, which suggested local thyroid cell stimulation by TSHR antibodies (35).
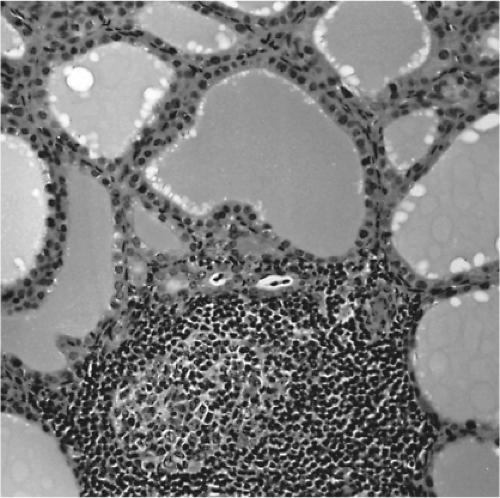 Figure 18A.6 Histologic section from a patient with Graves’ disease shows the patchy lymphocytic infiltrate that is characteristic of the disease. |
Functional Analysis of Intrathyroidal T Cells
As would be expected, activated B cells and T cells are more frequent in intrathyroidal lymphocyte cultures than peripheral blood cultures. T cells in patients with AITD are reactive to processed thyroid antigens (as peptides) (39). Such activated T cells enhance antibody (anti-Tg, anti-TPO, and TSHR antibody) secretion and have helper and regulatory functions. However, only 10% of activated T cells infiltrating the thyroid gland in patients with AITD proliferate in response to thyroid cell antigens indicating that “by-stander activation” (see later) is responsible for the majority of the cells. In Graves’ disease, intrathyroidal T-cell clones, when grown under appropriate conditions, are primarily Th2 with considerable T-cell helper activity (40,41). This supports the concept that the functional role of T cells in Graves’ disease is primarily a helper role.
T-cell Selection
As discussed above, positive and negative selection of T cells and B cells occurs in the thymus and/or in the peripheral
immune system. The TSH receptor, thyroglobulin, and TPO have been shown to be expressed by thymic epithelial cells and the expression of many autoantigens is under the control of the autoimmune regulator (AIRE) gene (42,43). Thyroid antigen reactive T cells should, in principal, therefore, be removed or anergized within the thymus, and if they escape deletion at that site they should normally be deleted by peripheral mechanisms. Anergy occurs when T cells and B cells bind antigen in the absence of second signals, as also discussed earlier. The resulting anergized T cells have potent suppressive actions and may act as regulatory cells but are also subject to apoptosis.
immune system. The TSH receptor, thyroglobulin, and TPO have been shown to be expressed by thymic epithelial cells and the expression of many autoantigens is under the control of the autoimmune regulator (AIRE) gene (42,43). Thyroid antigen reactive T cells should, in principal, therefore, be removed or anergized within the thymus, and if they escape deletion at that site they should normally be deleted by peripheral mechanisms. Anergy occurs when T cells and B cells bind antigen in the absence of second signals, as also discussed earlier. The resulting anergized T cells have potent suppressive actions and may act as regulatory cells but are also subject to apoptosis.
Apoptosis in Graves’ Disease
Autoimmune disease is a reflection of the failure to delete, suppress, or anergize antigen-specific T cells. In Graves’ disease there is a failure to achieve lasting tolerance to the TSH receptor antigen. One mechanism for deleting T cells is achieved via apoptosis and there are a number of pathways which signal this mechanism, i.e., by activation of T-cell surface molecules such as Fas antigen. Signaling is initiated by binding of Fas ligand expressed by thymic epithelium and thyrocytes may be involved in this pathway (see later) (44). There is also evidence that thyroid cells may become apoptosed even in hypertrophied Graves’ thyroid glands (36,45). This is a minor population of cells and may be related to the action of neutral TSHR-Abs (27). Apoptosed cells and the DNA released from such dead cells may also have a marked stimulatory influence on the autoimmune response; so this phenomenon should not be ignored (46).
The Intrathyroidal T-cell Receptor V Gene Repertoire
Most T-cell antigen receptors on the surface of T cells consist of two non-covalently linked chains (α and β), each with variable (V), diversity (D) (mainly β), and junctional (J) regions with common constant (Cα and Cβ) regions (47,48). Other T cells of less certain function have γ/d receptors. The V, D, and J genes code for recognition of the antigen–HLA complex by the T-cell receptor, affording antigen specificity. In addition to the many V (>100) and J (>50) genes present in the genome, random nucleotide (N) additions and deletions to the D region add immense complexity to the T-cell antigen receptor repertoire, causing this region, referred to as the third complementarity determining region (CDR3), to be of prime importance in antigen recognition (49). Most studies of intrathyroidal T cells have demonstrated bias in V gene utilization by T cells from within the thyroid as compared with peripheral blood from the same patient (8,50). Evidence has also been sought for clonal expansion of T-cell populations within the thyroid gland of patients by direct sequencing of the CDR3 regions of the T-cell antigen receptors of intrathyroidal T cells generated by RT-PCR. The most prominent V gene families were indeed representative of clonally expanded T cells, based on the evidence of multiple identical sequences within the generated fragments (51). Such information once again supports the concept of pauciclonal intrathyroidal T cells in Graves’ disease and points to the importance of T cells in disease etiology. Identical TcRs have also been shown to occur within the thyroid and retro-orbital tissues of patients with Graves’ disease and orbitopathy (50). A similar T-cell receptor bias has been observed in many other autoimmune diseases including rheumatoid arthritis and multiple sclerosis. Highly restricted T-cell responses occur early in autoimmune disease, but as the pathologic process progresses the response becomes less restricted, secondary to “determinant spreading” and bystander activation (see later) (52,53).
Regulatory Effects of T Cells in Autoimmune Thyroid Disease
With the identification of the CD4+/CD25+/Foxp3+ T-cell subset as a major regulatory cell population (54), these cells have been reported to be abnormal in some immune diseases including experimental autoimmune thyroiditis (55). However, their importance in human autoimmune thyroid disease remains uncertain, with limited evidence for and against a role in Graves’ disease (56,57). Early data also demonstrated that certain HLA-DR haplotypes conferred a reduced nonspecific regulatory T-cell function. For example, normal subjects with HLA-DR3 may have reduced suppressor T-cell activity, as compared with non-DR3 subjects. In addition, other mechanisms are in place to maintain thyroid-specific tolerance as discussed earlier:
The secretion of inhibitory cytokines by inflammatory immune cells,
The induction of anergy in T cells because of the absence of second signals,
The induction of apoptosis leading to immune cell deletion.
The Th17 Cells
This third set of helper T cells produce IL-21 and IL-22 as well as IL-17. These are highly pro-inflammatory cells that can trigger T- and B-cell proliferation and induce chronic inflammation and autoimmune disease in animal models (58). It is unlikely that such cells play a major role in Graves’ disease although some data suggest they may be important in Hashimoto’s thyroiditis (59,60).
Related posts:
 Anatomy and Pathology of the Thyrotrophs
Anatomy and Pathology of the Thyrotrophs
 Nonthyroidal Illness Syndrome
Nonthyroidal Illness Syndrome
 Toxic Adenoma and Toxic Multinodular Goiter
Toxic Adenoma and Toxic Multinodular Goiter
 Thyroid Hormones in Thermogenesis, Intermediary Metabolism, and Obesity
Thyroid Hormones in Thermogenesis, Intermediary Metabolism, and Obesity
 Overview of the Clinical Manifestations of Hypothyroidism
Overview of the Clinical Manifestations of Hypothyroidism
 Hypothyroidism in Infants and Children: Neonatal Screening
Hypothyroidism in Infants and Children: Neonatal Screening
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


