Plaque rupture is defined by fibrous cap disruption or fracture whereby the overlying thrombus is in continuity with the underlying necrotic core (4).
Plaque erosion is identified when serial sectioning through a thrombus fails to show communication with a necrotic core or deep intima; the endothelium is absent and the thrombus is superimposed on a plaque substrate primarily composed of smooth muscle cells (SMCs) and proteoglycans (5).
Calcified nodule is characterized by eruptive, dense calcified bodies protruding into the luminal space and represents the least frequent morphology associated with luminal thrombosis (3).
debatable, studies suggest that the loss of SMCs (death by apoptosis) may be involved as their remnant basement membranes can be visualized by periodic acid-Schiff (PAS) staining. Another feature of these lesions is microcalcification as prominently seen with anionic stains such as von Kossa’s method. These cell remnants and calcium apatite crystals can be observed by transmission electron microscopy as well. Calcium deposits may also be visualized on routine hematoxylin and eosin stains, provided the arteries have not undergone decalcification processes.
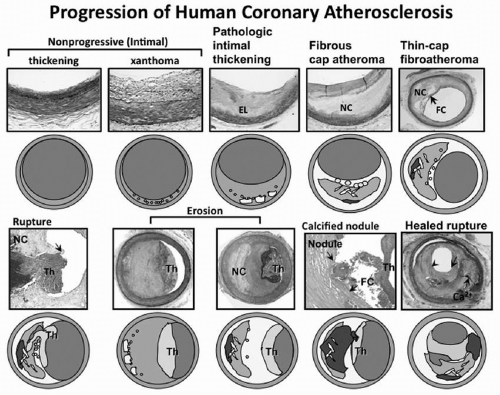 FIGURE 3.1 Spectrum of representative coronary lesion morphologies seen in our sudden coronary death population forming the basis for our modified AHA descriptive classification. The two nonprogressive lesions are intimal thickening and intimal xanthomas (foam cell collections known as fatty streaks, AHA type II). PIT (AHA type III transitional lesions) marks the first of the progressive plaques since they are the assumed precursors to more advanced FA. TCFAs are considered precursors to plaque rupture. Essentially missing from the AHA consensus classification are two alternative entities that give rise to coronary thrombosis, namely erosion and the calcified nodule. Erosions can occur on a substrate of PIT or fibroatheroma while calcified nodules (a minor but viable mechanisms of thrombosis) depict eruptive fragments of calcium that protrude into the lumen causing a thrombotic event. Lastly, healed plaque ruptures are lesions with generally smaller necrotic cores and focal areas of calcification where the surface generally shows areas of healing rich in proteoglycans. Multiple healed plaque ruptures are thought responsible for progressive luminal narrowing. AHA, American Heart Association; PIT, pathologic intimal thickening; FA, fibroatheromas; TCFAs, thin-cap fibroatheromas. (Reproduced with permission from Virmani R, Kolodgie FD, Burke AP, et al. Lessons from sudden coronary death: a comprehensive morphological classification scheme for atherosclerotic lesions. Arterioscler Thromb Vasc Biol. 2000;20:1262-1275.) (see color insert.) |
formation. This together with the death of macrophages in the setting of defective phagocytic clearance of apoptosis cells is thought to contribute to the development of late plaque necrosis.
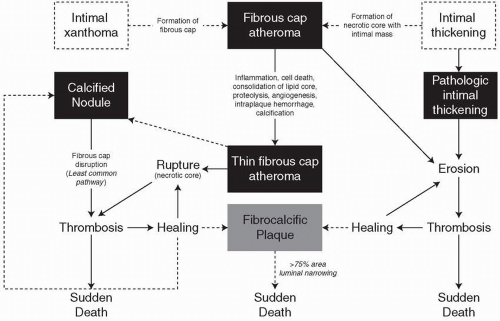 FIGURE 3.2 Simplified scheme for classifying atherosclerotic lesions modified from the current AHA recommendations. The boxed areas represent the seven categories of lesion. Dashed lines are used for two categories because there is controversy over the role that each plays in the initial phase of lesion formation, and both “lesions” can exist without progressing to a fibrous cap atheroma (i.e., AHA type IV lesion). The processes responsible for progression are listed between categories. Lines (solid and dotted, the latter representing the least-established processes) depict current concepts of how one category may progress to another, with the thickness of the line representing the strength of supportive evidence that the events occur. AHA, American Heart Association. (Reproduced with permission from Virmani R, Kolodgie FD, Burke AP, et al. Lessons from sudden coronary death: a comprehensive morphological classification scheme for atherosclerotic lesions. Arterioscler Thromb Vasc Biol. 2000;20:1262-1275.) |
mice subjected to medullar aplasia by radiation were given bone marrow-derived cells from Mfge8-deficient mice leading to the substantial accumulation of apoptotic debris within the developing lesion. Accumulated apoptotic material was associated with a marked acceleration of atherosclerosis accompanied by decreased anti-inflammatory (IL-10) production by protective T cells. In addition to lactadherin, other putative molecules in plaques potentially linked to efferocytosis and defective clearance of apoptotic debris include Fas ligand (18) and transglutaminase-2 (TG2) (19). Taken together, these correlative studies provide potential evidence for the involvement of key regulatory components in necrotic core expansion and further lesion development.
TABLE 3.1 MODIFIED AHA CONSENSUS CLASSIFICATION BASED ON MORPHOLOGIC DESCRIPTION | ||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||
Related posts:
 Use of Noninvasive Methods to Diagnose Cardiovascular Disease in Dyslipidemic Patients
Use of Noninvasive Methods to Diagnose Cardiovascular Disease in Dyslipidemic Patients
 Dyslipidemia in Children and Adolescents
Dyslipidemia in Children and Adolescents
 Dietary Treatment of Dyslipidemia
Dietary Treatment of Dyslipidemia
 Disorders of Hypertriglyceridemia
Disorders of Hypertriglyceridemia
 Dyslipidemia in the Elderly: Special Insights to Inform the Management Strategy
Dyslipidemia in the Elderly: Special Insights to Inform the Management Strategy
 Trans Fatty Acids, Dyslipidemia, and Cardiovascular Disease
Trans Fatty Acids, Dyslipidemia, and Cardiovascular Disease
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


