PARATHYROID HORMONE RESISTANCE
PSEUDOHYPOPARATHYROIDISM TYPE 1
The term pseudohypoparathyroidism (PHP) describes a heterogeneous syndrome characterized by biochemical hypoparathyroidism (i.e., hypocalcemia and hyperphosphatemia), increased plasma levels of PTH, and peripheral unresponsiveness to the biologic actions of PTH. Thus PHP differs substantially from true hypoparathyroidism; in contrast with the latter condition, PTH secretion is excessive and the parathyroid glands are hyperplastic.105
PHP was the first recognized human disease to be ascribed to diminished responsiveness to a hormone by otherwise normal target organs (subsequently, many others have been described) (see below).106 Albright’s initial description of the blunted calcemic and phosphaturic response to PTH administration in patients with PHP provided the basis for his original hypothesis that the disorder is due to target organ resistance.107
MOLECULAR BASIS FOR PSEUDOHYPOPARATHYROIDISM: A MODEL FOR HUMAN G PROTEIN DISEASE
Characterization of the molecular basis for PHP commenced when research showed that cyclic adenosine monophosphate (cAMP) mediates the actions of PTH on kidney and bone and that PTH infusion in humans leads to a significant increase in urinary excretion of cAMP.108,109 When these observations were applied to the study of PHP, affected persons were found to have a blunted urinary response of nephrogenous cAMP to PTH infusion in comparison with normal persons and patients with other forms of hypoparathyroidism (Fig. 60-4).109 These findings formed the basis for the most reliable test presently available for the diagnosis of PHP type 1. Moreover, these results suggested that PTH resistance is caused by a defect in the plasma membrane–bound PTH receptor–adenylate cyclase complex that produces cAMP. Evidence
is accumulating that some actions of PTH may be cAMP independent, with signal transduction through inositol phospholipids, intracellular calcium mobilization, and protein kinase C activation (Fig. 60-5).110,111,112 and 113 This may explain why some patients with PHP who have blunted urinary cAMP responses after PTH infusion still have evidence for PTH-mediated effects on renal tubular calcium handling114,115 and skeletal remodeling (see later).
is accumulating that some actions of PTH may be cAMP independent, with signal transduction through inositol phospholipids, intracellular calcium mobilization, and protein kinase C activation (Fig. 60-5).110,111,112 and 113 This may explain why some patients with PHP who have blunted urinary cAMP responses after PTH infusion still have evidence for PTH-mediated effects on renal tubular calcium handling114,115 and skeletal remodeling (see later).
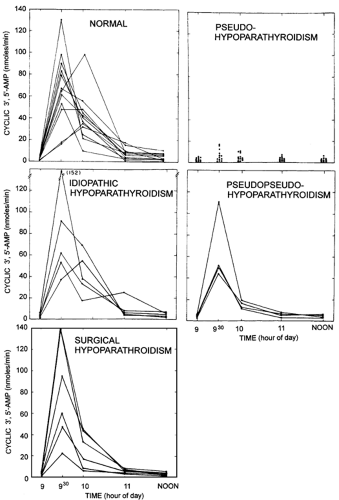 FIGURE 60-4. Cyclic adenosine monophosphate (AMP) excretion in urine in response to the injection of parathyroid hormone (300 USP units of bovine parathyroid extract infused from 9:00 to 9:15 a.m.). (Reproduced from Chase LR, Melson GL, Aurbach GD. Pseudohypo-parathyroidism: defective excretion of 3′5′-AMP in response to parathyroid hormone. J Clin Invest 1969; 48:1836, by permission of the authors and the American Society for Clinical Investigation.) |
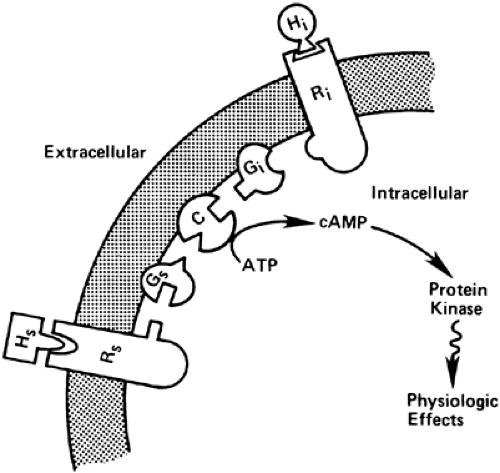 FIGURE 60-5. Schematic outline of the adenylate cyclase–cyclic adenosine monophosphate (cAMP) system. Hs and Hi denote stimulatory and inhibitory agents, respectively; Rs and Ri stimulatory and inhibitory receptors; and Gs and Gi the stimulatory and inhibitory guanine nucleotide–binding regulatory proteins. The rate of conversion of substrate adenosine triphosphate (ATP) to product cAMP by the catalytic (C) unit of adenylate cyclase is regulated by the interactions of Gs and Gi. Details of the interactions are described in the text. |
The adenylate cyclase system is far more complex than originally suspected, consisting of at least three types of proteins embedded in the plasma membrane (i.e., receptors, G proteins, and adenylate cyclase [Fig. 60-6; see Chap. 4]).116 The signal-transducing G proteins are comprised of three subunits (αβγ) that are encoded by 16α, 6β, and 12γ genes. The α subunits are loosely associated with tightly coupled βγ dimers; this facilitates great combinatorial variability and allows G proteins to interact with as many as 1000 different GPCRs and effector proteins.117 G proteins have been divided into four subfamilies: Gs, Gi, Gq, and G12, based on structural and functional similarities. Members of the G s family activate adenylate cyclase and members of the Gq family activate phospholipase C; members of the Gi subfamily inhibit adenylate cyclase (Gi) or participate in visual (Gt1 and Gt2) or gustatory (Ggust) neurosensory processes. G13 stimulates the exchange of sodium and hydrogen ions and cytoskeletal rearrangements.117,118 and 119 The activity of the heterotrimeric protein is regulated by the binding and hydrolysis of guanosine triphosphate (GTP) by the Gα subunit. When Gβ is bound to GDP, it is inactive and loosely associates with the βγ dimer. Receptor activation results in release of GDP by the heterotrimeric αβγ complex, binding of GTP to Gα, and dissociation of the Gα-GTP complex from the βγ dimer and receptor. Both the Gα-GTP chain and free βγ dimer can regulate downstream effectors. Termination of receptor signaling results from the hydrolysis of GTP to guanosine diphosphate (GDP), allowing the βγ dimer to reassociate with Gα-GDP.117
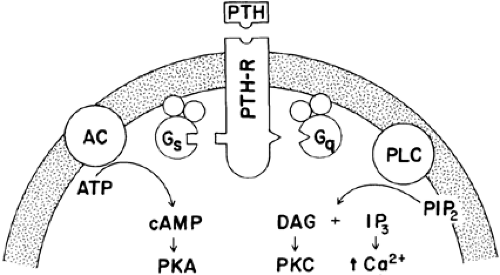 FIGURE 60-6. Cell surface receptors for parathyroid hormone (PTH) appear to be coupled to two classes of G proteins. Gs mediates stimulation of adenylate cyclase (AC) and the production of cyclic adenosine monophosphate (cAMP), which in turn activates protein kinase A (PKA). Gq stimulates phospholipase C (PLC) to form the second messengers inositol 1,4,5-trisphosphate (IP3) and diacylglycerol (DAG) from membrane-bound phosphatidylinositol 4,5-bisphosphate (PIP2). IP3 increases intracellular calcium (Ca2+) and DAG stimulates protein kinase C (PKC) activity. Each G protein consists of a unique α chain and a βγ dimer. (ATP, adenosine triphosphate; PTH-R, parathyroid hormone receptor.) (From Bilezikian JP, Levine MA, Marcus R, eds. The parathyroids: basic and clinical concepts. New York: Raven Press, 1994:782.) |
Mutations in G proteins tend to affect a variety of tissues, as demonstrated by the occurrence of multihormonal resistance in PHP type 1a. By contrast, receptor mutations are generally limited to one or a few tissues in which the defective receptor is expressed (e.g., defects in the CaR produce a disturbance in calcium sensing that appears limited to parathyroid and kidney cells). The determinants of phenotypic expression of the G protein and GPCR disease are the range of expression of the gene (tissue distribution), the developmental timing of the mutation (germline vs. somatic), and the nature of the mutation (loss or gain of function).
PSEUDOHYPOPARATHYROIDISM TYPE 1A
Albright’s original description of PHP focused on PTH resistance in this disorder. Resistance to PTH alone would be consistent with a defect in the cell surface receptor specific for PTH. However, some patients with PHP type 1 are resistant to multiple hormones whose effects are mediated by cAMP and have additional abnormalities, such as hypothyroidism and hypogonadism,120 mental retardation,121 and defective olfaction.122 These patients, whose disorder is referred to as PHP type 1a, have ˜ 50% reduction in the activity of Gs (Fig. 60-7). This reduction occurs in all tissues that have been examined (including erythrocytes, platelets, fibroblasts, transformed lymphocytes, and, in one case, renal cortex).123,124 and 125 A generalized deficiency of Gs activity apparently results in a reduced ability of hormones and neurotransmitters to activate adenylate cyclase in diverse tissues and thereby leads to widespread hormone resistance (Fig. 60-8).
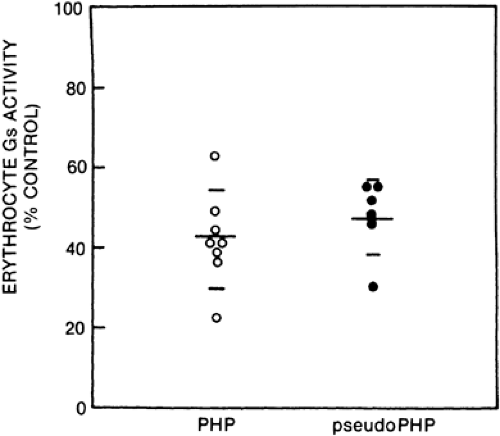 FIGURE 60-7. Gs activity in pseudohypoparathyroidism (PHP) type 1a and in pseudopseudohypoparathyroidism (pseudoPHP). Gs activity was measured in erythrocyte membrane extracts by complementation with Gs-deficient membranes from S49 cyc cells. The resultant adenylate cyclase activity is expressed as a percentage of a pooled normal human erythrocyte membrane standard. (From Levine MA, Jap T, Mauseth RS, et al. Activity of the stimulatory guanine nucleotide–binding protein is reduced in erythrocytes from patients with pseudohypoparathyroidism and pseudopseudohypoparathyroidism: biochemical, endocrine, and genetic analysis of Albrights hereditary osteodystrophy in six kindreds. J Clin Endocrinol Metab 1986; 62:497.) |
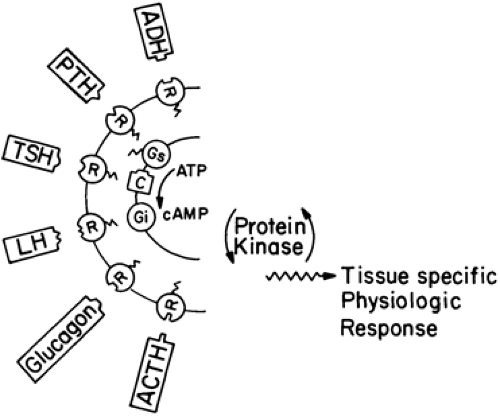 FIGURE 60-8. Model showing some of the multiple receptors that are coupled by means of Gs to activation of adenylate cyclase. Gene mutations that reduce Gs α activity commonly impair transmembrane signal transduction processes and result in hormone resistance. By contrast, activating mutations of Gsα produce constitutive signal transduction in the absence of hormone or neurotransmitters and can result in autonomous cell function and proliferation. (ADH, antidiuretic hormone; PTH, parathyroid hormone; TSH, thyroid-stimulating hormone; LH, luteinizing hormone; ACTH, adrenocorticotropic hormone; R, receptor; C, catalytic unit of adenylate cyclase; ATP, adenosine triphosphate; cAMP, cyclic adenosine monophosphate.) |
Patients with PHP type 1a show a unique phenotype characterized by round facies, short stature, obesity, brachydactyly (short metacarpal and metatarsal bones), heterotopic subcutaneous ossification, and bony exostoses (Fig. 60-9). Mental retardation may be present. In 1942, Albright and coworkers107 first observed these unusual developmental defects, subsequently termed Albright hereditary osteodystrophy (AHO), in three patients during the initial description of PHP.
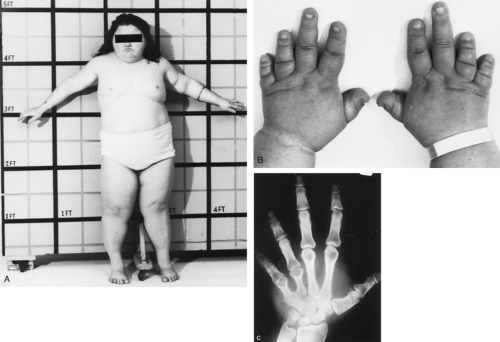 FIGURE 60-9. A, Young woman with Albright hereditary osteodystrophy. Note the short stature and rounded facies. B, Brachydactyly, particularly manifest in many patients as shortened fourth fingers. A “dimpling” of the fourth and fifth knuckle region may be seen when a fist is made. C, The radiograph reveals characteristic shortening of the fourth metacarpal, as well as shortening of the fifth metacarpal. |
Ten years after the description of PHP, Albright and colleagues126 described a patient with a habitus typical of AHO but who lacked biochemical evidence of target organ resistance to PTH. This normocalcemic variant of AHO was termed pseudopseudohypoparathyroidism (pseudoPHP) to call attention to the physical similarity with AHO yet indicate the metabolic dissimilarity (PTH responsiveness) of this disorder.
PseudoPHP is genetically related to PHP. Early clinical observations of AHO kindreds in which several affected members had only AHO (i.e., pseudoPHP), whereas others had PTH resistance as well (i.e., PHP) first suggested that the two disorders might reflect variability in expression of a single genetic lesion. Further support for this hypothesis derives from biochemical
studies which indicate that patients with pseudoPHP who are related to patients with PHP type 1a have equivalent functional Gsα deficiency (see Fig. 60-7)127 and identical gene defects (see later). Therefore, it seems reasonable to use the term Albright hereditary osteodystrophy to simplify description of this syndrome and to acknowledge the common clinical and biochemical characteristics that patients with PHP type 1a and pseudoPHP share.
studies which indicate that patients with pseudoPHP who are related to patients with PHP type 1a have equivalent functional Gsα deficiency (see Fig. 60-7)127 and identical gene defects (see later). Therefore, it seems reasonable to use the term Albright hereditary osteodystrophy to simplify description of this syndrome and to acknowledge the common clinical and biochemical characteristics that patients with PHP type 1a and pseudoPHP share.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


