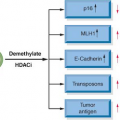
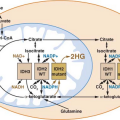
ductal carcinomas.17 The first genetic change in the ducts is probably not (or not always) a KRAS gene mutation, for the prevalence of this mutation is highest in the more advanced lesions (Table 17.1).18,19 KRAS is one of a family of RAS genes that can harbor mutations in human cancers. The other RAS genes include NRAS and HRAS, although it is possible that only KRAS is mutated in pancreatic carcinomas.
TABLE 17.1 GENETIC PROFILE OF PANCREATIC CARCINOMA | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
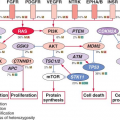
developed as drugs, they have not been successful anticancer agents. There are many reasons for this. Although Hras protein is linked predominantly through farnesyl groups, the Kras protein can be alternately prenylated by geranylgeranyl linkages. Unfortunately, the latter type of linkage is thought to be critical for a wider number of cellular proteins, and for fear of excessive toxicity, geranylgeranyl linkages have not usually been considered as an attractive drug target. Kras protein also appears to bind more tightly than Hras to the farnesyltransferase enzyme, requiring higher drug concentrations.16 Additionally, the artificial models usually employed the engineered overexpression of the Ras protein, a situation in which the unattached Ras proteins would serve as a dominant-negative inhibitor, binding the necessary interacting proteins and sequestering them in the cytoplasm to ensure the inactivation of all three Ras pathways. Such a concentrationdriven mechanism would presumably not occur under the normal levels of Ras proteins present in human cancers.20 Indeed, it is proposed that the limited efficacy of farnesyltransferase inhibitors observed in some experimental models and in clinical trials may be attributable to a cellular target not yet identified.21 Attention has turned to compounds that target the downstream mediators, such as Raf and Mek protein kinase inhibitors.
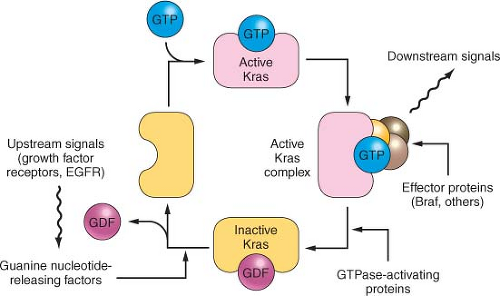 FIGURE 17.1 The KRAS pathway. KRAS normally integrates and regulates signals arising in the growth factor receptors that are passed to KRAS using the Grb2 and the Sos1 nucleotide exchange factor. The active GTP-bound form of KRAS recruits effector proteins such as Raf1 and Braf, in turn stimulating the downstream mitogen-activated protein kinases such as MEK and ERK and activating certain transcription factors. The EGF receptor can be overexpressed and occasionally mutated to provide inappropriately strong upstream signals, and the BRAF protein can be activated by point mutation, but more often in pancreatic cancer the Kras protein is mutated. These latter mutations impair the GAP (GTPase-activating protein)-stimulated reaction that normally returns Kras to the inactive state. |
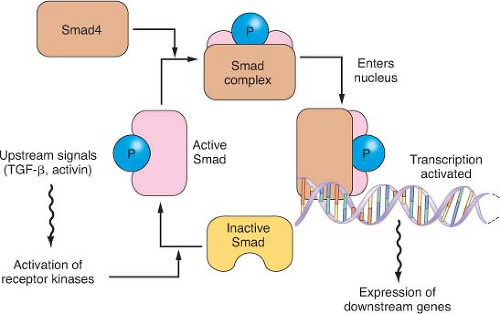 FIGURE 17.2 The transforming growth factor-beta (TGFβ)/Activin/Smad pathway. Dimeric kinase receptors of the TGFβ superfamily respond to extracellular ligands, causing phosphorylation of one or more of the receptor-associated Smad proteins and leading them to complex with the unphosphorylated common Smad, Smad4. This complex binds to specific DNA sequences and works with other transcription factors to stimulate gene expression. Mutations in pancreatic cancer can inactivate either partner of the dimeric receptors that respond to extracellular TGFβ or activin. More commonly, however, mutations and large deletions in the SMAD4 gene destabilize its protein product or ablate gene expression. |
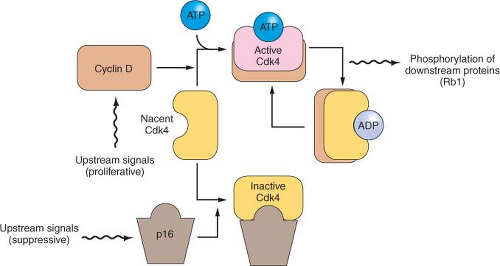 FIGURE 17.3 The p16/RB1 pathway. p16 binds to, inhibits, and thereby controls the availability of the cyclin-dependent kinases Cdk4 and Cdk6 (not shown). When activated by binding to cyclin D, these kinases phosphorylate and thereby inactivate the Rb1 tumor suppressor protein. The activity of p16 is controlled in a complex manner, through changes in gene expression and by displacement reactions involving other similar kinase inhibitor proteins. p16 mutations and deletions are nearly ubiquitous in pancreatic cancer, resulting in dysregulation of these cyclin-dependent kinases that regulate the cell division cycle. |
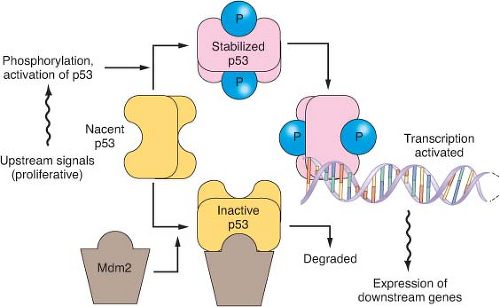 FIGURE 17.4 The p53 pathway. Many modes of control affect p53 activity, one of which is shown in the diagram. Stresses such as DNA damage result in phosphorylation of p53, preventing its degradation by an Mdm2-directed pathway. When stabilized, p53 binds to specific DNA sequences and activates the transcription of many genes, including Mdm2 as part of a negative feedback loop. When p53 is mutated, it fails to bind effectively to DNA to activate transcription. Because Mdm2 then lacks its transcriptional stimulus from p53, mutant but inactive p53 proteins are usually expressed at very high levels. |
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


