Other Tumors of Undefined Neoplastic Nature
Sean V. McGarry
C. Parker Gibbs
Langerhans cell histiocytosis (LCH) and Erdheim-Chester disease (ECD) are histiocytoses: rare benign lesions of bone in which the proliferating tumor cell of origin is a histiocyte. There are two types of histiocytes: (1) macrophages arising from monocytes and (2) dendritic cells arising from Langerhans cells. In LCH the involved cell is a Langerhans cell, in ECD a macrophage. Histiocytoses in general exist in multiple forms, ranging from a solitary lesion to a diffuse systemic disease, and they can affect any organ or tissue in the body. This section deals with the two histiocytoses involving bone: LCH and ECD. LCH is most commonly a solitary lesion occurring in children and young adults and often requires surgical intervention. ECD is often a systemic disease of older adults and very rarely involves surgical intervention.
Erdheim-Chester Disease
The original description of ECD was made by Chester in 1930. In 1972, Jaffe first used the eponym ECD to recognize that description. In 1983, a review of the literature by Alper found that 47 cases had been described up to that point. ECD remains an extremely rare entity; thus, our knowledge of the natural history of the disease and its treatment is somewhat limited. Its etiology is unknown.
Pathogenesis
Epidemiology
Typically a disease of older adults (Fig. 5.8-1)
Age range 7 to 84 years
Mean age 53 years
Slight male predominance
Affects long bones most commonly
Bilateral and symmetric
Classically affects diaphysis and metaphysis, sparing epiphysis (not always)
Lower extremity (femur, tibia, fibula) more common than upper extremity (humerus, radius, ulna)
Usually spares flat bones
Pathophysiology
Systemic lipid storage disorder affecting histiocytes
Lipid-laden histiocytes form granulomas in bones, other organs or tissues.
Prognosis and Natural History
Because of the infrequency of the disease, little is known about prognostic factors.
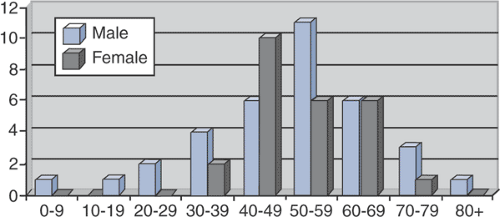
Figure 5.8-1 Sex and age at time of diagnosis for 59 patients with Erdheim-Chester disease. (After Veyssier-Belot C, Cacoub P, Caparros-Lefebvre D, et al. Erdheim-Chester disease: Clinical and radiologic characteristics of 59 cases. Medicine 1996;75:157–169.)
Prognosis is related to extent of visceral involvement.
In some patients the disease is quiescent.
In most patients the disease is progressive, and patients die of end-organ failure
Respiratory distress or pulmonary fibrosis
Heart failure
Renal failure
Classification
There currently is no accepted classification scheme for ECD.
Diagnosis
Physical Examination and History
A definitive diagnosis requires the combination of history, physical examination findings, radiographs, and a tissue diagnosis.
Classical radiographic findings with bilateral, symmetric osteosclerosis of the diaphysis and metaphysis with epiphyseal sparing can be considered pathognomonic.
Radiologic Features
Densely sclerotic changes of the metaphysis and diaphysis, with coarsened trabeculae
Typically spares the epiphysis
Rarely, lesions are more lytic in nature.
Long bones most commonly affected (Fig. 5.8-3)
Lower extremities more commonly affected than upper extremities
Table 5.8-1 Clinical Manifestations of Erdheim-Chester Disease According to Organ of Involvement | ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||
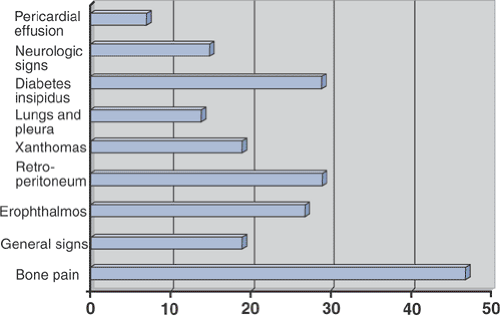 Figure 5.8-2 Prevalence of clinical signs in 59 patients with Erdheim-Chester disease. (After Veyssier-Belot C, Cacoub P, Caparros-Lefebvre D, et al. Erdheim-Chester disease: Clinical and radiologic characteristics of 59 cases. Medicine 1996;75:157–169.) |
Diagnostic Workup Algorithm
Radiographs are nearly always pathognomonic for ECD.
When seen in concert with typical complaints of bone pain, exophthalmos, diabetes insipidus, or xanthomas, these patients are not a diagnostic challenge.
They can show elevated erythrocyte sedimentation rate, alkaline phosphatase, or serum lipid profiles.
However, laboratory testing is unreliable and nonspecific.
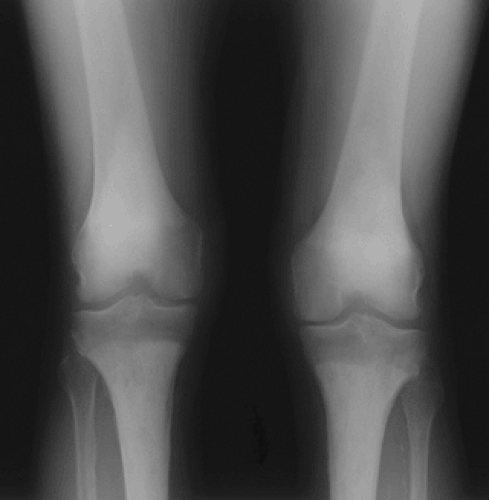
Figure 5.8-3 Radiograph of patient with Erdheim-Chester disease showing typical radiographic changes.
If there is any question as to the diagnosis, a biopsy of an accessible skeletal lesion is performed.
Histopathologic evaluation demonstrates infiltration of the bone by foamy, lipid-laden histiocytes, with giant cells and rare lymphocytes.
Immunostaining demonstrates negative staining for both S-100 and CD1a (which would suggest the diagnosis of LCH).
Treatment
Surgical and Nonoperative Options
Occasional need for biopsy to confirm diagnosis
No need for surgical intervention other than rare prophylactic fixation to prevent fracture
No good nonoperative treatment options, but historically patients are treated with any combination of the following:
Steroids
Chemotherapy
Radiation
Results and Outcome
Overall Outcome
With no good treatment options for ECD, overall survival is rather bleak. In a review of 37 patients followed for an average of 30 months, Veyssier-Belot reported a mortality rate of 59%. Other reports suggest a mortality rate of approximately 33%, but with limited follow-up. Patients die of end-organ failure (pulmonary fibrosis, heart failure, or renal failure most commonly).
Specific Treatment Outcome
Because there is not a consensus as to the appropriate treatment of ECD in combination with the rarity of the diagnosis, there are not sufficient numbers to assess specific treatment outcomes. In the review by Veyssier-Belot, the following results are presented:
Steroid therapy alone in 12 patients
Effective transiently in 4 patients
Ineffective in 8
Chemotherapy in combination with steroids in 8 patients
4 patients improved
No effect in 4 patients
Radiation therapy in 6 patients
Transiently effective in 3 for bone pain
Not effective in 3 to treat exophthalmos
Langerhans Cell Histiocytosis
In 1953, Lichtenstein, noting the histological similarities in eosinophilic granuloma, Hand-Christian-Schüller, and Letterer-Siwe, coined the term “histiocytosis X” as a general term to encompass all three diagnoses. We know today that all three diseases originate from a Langerhans cell and refer to the group of disorders as the Langerhans cell histiocytoses (LCH) or granulomatoses. The three share a common histopathology and are considered to be clinical variations of the same disease.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


