Osteosarcoma
Francis R. Patterson
Osteosarcoma is the most common sarcoma of bone in young people. While relatively rare compared to other orthopaedic disorders, an appropriate level of suspicion must be maintained for osteosarcoma as a source of pain in this age group. When diagnosed early and treated appropriately, survival with a highly functional limb can often be achieved in this age group. Most osteosarcoma patients present with pain as the initial symptom, but some patients first notice this after an injury or falsely attribute their symptoms to a minor trauma. Therefore, pain that does not improve in the expected time period, or pain that is worsening despite treatment or rest, should raise a red flag, and an appropriate work-up, starting with plain radiographs, must be pursued. Osteosarcoma has a bimodal distribution with a second but smaller peak in late adulthood. Adult osteosarcomas are often secondary to conditions such as Paget’s disease or prior radiation (Table 6.1-1). After biopsy, the standard treatment of osteosarcoma includes preoperative chemotherapy, followed by resection, and postoperative chemotherapy when appropriate.
Pathogenesis
Etiology
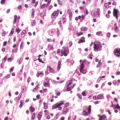
Osteosarcoma is a tumor composed of a malignant spindle cell stroma (background) with malignant osteoblasts
that produce tumor osteoid (collagenous immature bone that appears pink on hematoxylin-and-eosin [H&E] staining) or bone (Fig. 6.1-1).
Table 6.6-1 Comparison of Vascular Sarcomas of Bone
Sarcoma Type
Age
Gender
Anatomic Distribution
Gross Appearance
Histologic Features
Other Details
Hemangioendothelioma
First through ninth decades
M > F (slightly)
Half involve lower extremity; any bone affected
Firm, friable, bloody
Well-formed vascular spaces (“staghorn spaces”) lined by plump endothelial cells.
Intermixed with corded pattern mimicking carcinoma.
Multicentricity common; one third of cases multifocal
Epithelioid hemangioendothelioma
Second through eighth decades; peaks in second and third decades
M > F (slightly)
Femur most common; any bone affected
Firm, lobulated, tan
Corded, nested, or stranded pattern of plump endothelial cells with eosinophilic cytoplasm within hyalinized stroma (see Fig. 6.6-1).
May form narrow vascular channels.
Occasional cytoplasmic vacuoles (represent primitive blood vessel lumina).
Signet ring-like appearance.
Angiosarcoma
Peaks in fourth decade
M > F (slightly)
Long tubular bones and spine most common; any bone affected
Firm, bloody
Less vasoformative than hemangioendotheliomas.
Regions with vascular differentiation and plump malignant endothelial cells.
Some areas may show epithelioid appearance.
As is the case with most sporadic occurring malignancies, the factors that lead to the development of an osteosarcoma are largely unknown.
Most likely, a mutation or group of mutations occurs that leads to uncontrolled growth of the mutant cells.
Evidence exists to support the role of genetic abnormalities in patients with osteosarcoma, though these abnormalities are not identified in most patients.
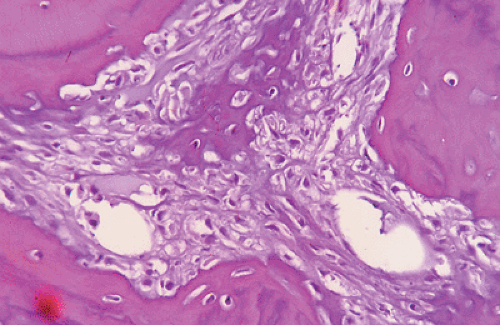
Figure 6.1-1 H&E stain shows a malignant spindle cell (osteoblast) stroma with lace-like (pink) osteoid and calcified (darker) osteoid in center, typical of a high-grade osteosarcoma.
Two tumor suppressor genes may play a significant part in tumorigenesis in osteosarcoma: p53 (chromosome 17) and Rb (chromosome 13).
Reported familial patterns of osteosarcoma
Chromosome 13:14 rearrangement in sisters
Deletion of part of chromosome 13 resulting in inactivation of the retinoblastoma (RB) gene in cousins
Two genetic conditions that predispose to development of osteosarcoma
Retinoblastoma patients
May have germline mutation of Rb gene
Increased risk of osteosarcoma
Li-Fraumeni syndrome (a familial cancer syndrome; p53 gene abnormalities)
Increased risk of osteosarcomas and other malignancies
Mothers of children with sarcomas have up to a three times increased risk of breast carcinoma.
Epidemiology
Osteosarcoma is the most common type of bone sarcoma. It is neither the most common primary bone malignancy nor the most common malignancy affecting bone. The most common primary bone malignancy is multiple myeloma, and the most common malignancy affecting bone is metastatic carcinoma.
Bimodal peak age incidence
Crude incidence: 0.3 per 100,000 per year in United States (roughly 900 per year)
Majority occur within the second decade (∼60%)
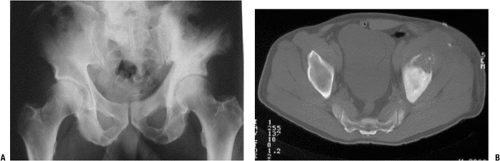
Figure 6.1-2 (A) Plain radiograph shows an area of bony destruction in the anterior left iliac wing and pagetic changes throughout the left hemipelvis. (B) Axial CT image shows lesion with soft tissue extension consistent with an osteosarcoma secondary to Paget’s disease of the pelvis.
Most occur at age <30 (∼85%)
Second peak age >55; often secondary osteosarcomas (e.g., Paget’s sarcoma or postradiation sarcoma)
Classic secondary osteosarcomas represent 5% to 7% of all osteosarcomas; usually with worse prognosis.
Definition: osteosarcomas that occur in relation to previous exposures or procedures as well as in the presence of other primary diseases
Paget’s osteosarcoma (Fig. 6.1-2)
Most common of the “secondary osteosarcomas”
Estimated 1% of Paget’s patients may develop osteosarcoma
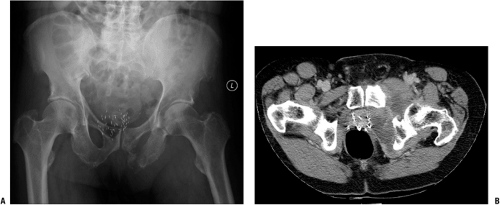
Figure 6.1-3 (A) Plain anteroposterior radiograph of the pelvis shows a destructive lesion of the left inferior pubic ramus. Note the metallic seeds previously placed for treatment of prostate cancer in combination with external-beam radiation 8 years prior. (B) Axial CT image shows bony destruction and soft tissue mass extending from pubic ramus. With the patient’s history, biopsy confirmed the diagnosis of postradiation osteosarcoma.
Majority in polyostotic Paget’s, although they do occur in monostotic disease
Reports as high as 5% of polyostotic symptomatic patients
Postradiation osteosarcomas occur in bone that is in a previously irradiated field (Fig. 6.1-3).
Usually >3 years after exposure
May occur decades after treatment for other malignancies or after high-dose exposure
Disease-associated osteosarcomas
Pathophysiology
Local Growth
Osteosarcomas usually arise within the metaphyseal region of long bones. They may be located within the bone or on the surface of the bone. If untreated, osteosarcomas will continue to grow, with local destruction of bone and extension outside the bone into the surrounding soft tissues. The physis and articular cartilage may act as a relative barrier to tumor extension, but epiphyseal or intra-articular extension is still seen frequently.
Metastases
Osteosarcomas, as with all sarcomas, usually metastasize hematogenously.
Lymph node metastases are not common and usually present only very late in the course of metastatic disease.
15% to 20% of patients present with metastases at time of diagnosis.
Most common site of metastasis: lungs (Fig. 6.1-4)
Second most common: bone
Skip lesions: distinct smaller areas apart from primary tumor within same bone
Prognosis: same as or worse than distant metastases (lung or bone)
Death
Occurs most commonly due to respiratory failure secondary to widespread pulmonary metastases, but also due to other sequelae of tumor burden
Superior vena cava obstruction
Pneumonia
Hemorrhage into tumor
Sepsis
Chemotoxicity (1% to 5%)
Classification
Osteosarcoma and Variants
“Classic” osteosarcoma, also referred to as conventional osteosarcoma, and central high-grade osteosarcoma represent the majority of osteosarcomas (up to 75% in some series). Variants of osteosarcomas exist that present differently, have unique radiographic and histologic characteristics, may be treated differently, and may convey a worse or better prognosis than conventional osteosarcoma. It is important that these differences are identified and understood so that correct diagnosis and treatment may be rendered.
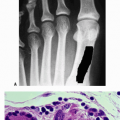
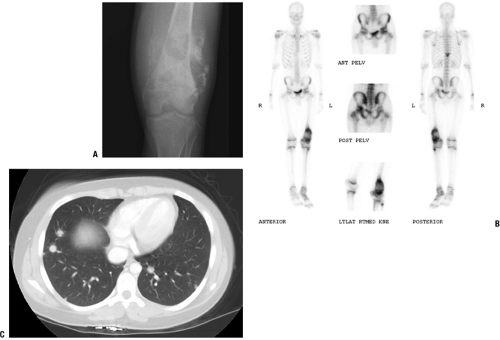 Figure 6.1-4 A 16-year-old boy with distal femoral lesion. (A) Plain radiograph shows abundant bone production and soft tissue extension strongly suggestive of an osteosarcoma. (B) Whole-body Tc-99 bone scan reveals a lesion of the ipsilateral acetabulum and thoracic spine. (C) CT axial image of the chest also reveals multiple pulmonary metastases. |
Pathology
Osteosarcoma is classically described as a high-grade spindle cell sarcomatous stroma with malignant osteoblasts that produce malignant osteoid or bone. The tumor cells are typically anaplastic (less differentiated), may show marked atypia and pleomorphic (widely variable) nuclei, and may show many and/or bizarre mitoses. There may be areas of osteoblastic (osseous), fibroblastic (fibrous), or chondroblastic (cartilage) appearance, but if there is the presence of malignant osteoid (wavy, lace-like, uncalcified bone matrix produced by malignant osteoblasts), the diagnosis of osteosarcoma is made regardless of the associated areas.
Grade
The grade of an osteosarcoma is used to:
Plan treatment: low-grade sarcomas are not treated with chemotherapy
Predict prognosis: low-grade osteosarcomas are less likely to develop metastases
Most osteosarcomas are high-grade tumors.
Low- and intermediate-grade osteosarcoma variants do exist (Box 6.1-1).Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree