53.1
Introduction
The systemic and focal joint inflammation that characterizes many chronic inflammatory diseases of autoimmune origin, or rheumatologic diseases, is frequently accompanied by adverse effects on the skeleton. Much of the attention has focused on the focal bone resorption in articular and periarticular bone associated with rheumatologic inflammation and reduced bone formation, as seen in rheumatoid arthritis (RA), the prototypical inflammatory joint disease. However, it is clear that in many of the rheumatic diseases there is also an effect on systemic bone remodeling, as numerous studies have identified osteoporosis and increased risk of fracture as substantial contributors to disease morbidity. In part, these adverse skeletal effects may be related to therapies used for treatment. This review will focus on the inflammatory rheumatologic disorders that target the articular and periarticular tissues, including RA, juvenile idiopathic arthritis (JIA) (previously classified as juvenile RA), the seronegative spondyloarthropathies, and systemic lupus erythematosus (SLE). It is important to note, that many of the related immune-mediated rheumatic conditions, such as systemic vasculitis, may also be accompanied by adverse effects on skeletal remodeling and increased risk for osteoporosis, in part related to the use of high-dose glucocorticoids and other therapies that adversely affect the skeleton.
53.2
Rheumatoid arthritis
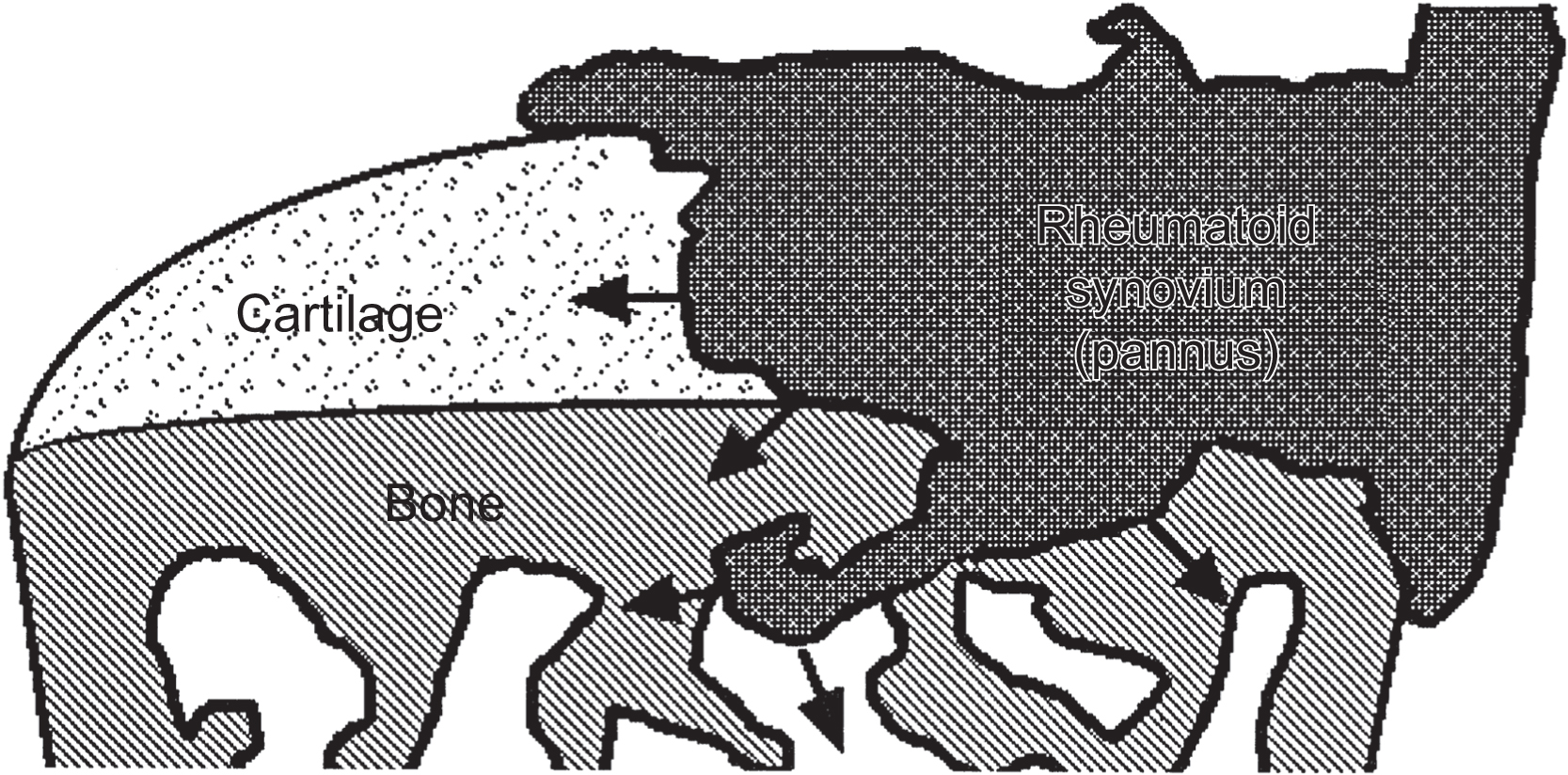
RA represents an excellent model for gaining insights into the effects of local as well as systemic consequences of inflammatory processes on skeletal tissue remodeling. RA is a relatively common systemic inflammatory disorder affecting between 1% and 2% of the adult population throughout the world. The etiology of RA is unknown, but there is considerable evidence that both genetic and environmental factors are involved in its pathogenesis . The hallmark of RA is the development of a chronic inflammatory process that targets the synovial lining of diarthrodial joints, causing synovial hyperplasia, and resulting in articular joint damage consisting of subchondral cysts, sclerosis, erosive bone damage, and cartilage loss. The earliest change is the synovial hyperplasia, containing a heterogeneous population of macrophage-like cells (A cells) and synovial fibroblasts (B cells). This process is accompanied by endothelial cell proliferation with neovascularization and perivascular infiltration with lymphocytes, plasma cells, and activated macrophages . As the disease progresses, the inflamed synovial tissue migrates over the articular surface, forming a pannus ( Fig. 53.1 ). At the interface between the pannus and hyaline cartilage, there is focal destruction of the cartilage matrix, and at site of contact between the inflamed synovium and the bone surface, there is osteoclastic bone resorption with resulting erosion ( Fig. 53.2 ). In addition to these focal bone changes, there is generalized loss of periarticular bone in these inflamed joints, also mediated by increased osteoclast activity .
Although the focal joint pathology accounts for a major component of the disease morbidity, RA is clearly a systemic illness that can produce adverse effects on extraarticular organs and is often accompanied by generalized systemic features of inflammation. This inflammation may affect the entire skeleton, resulting in progressive loss of bone mass and increased risk for fracture . The skeletal effects result in a higher prevalence of osteoporosis among RA patients than matched controls, and an increased fracture risk at both vertebral and appendicular sites .
53.3
Focal subchondral and marginal bone erosions
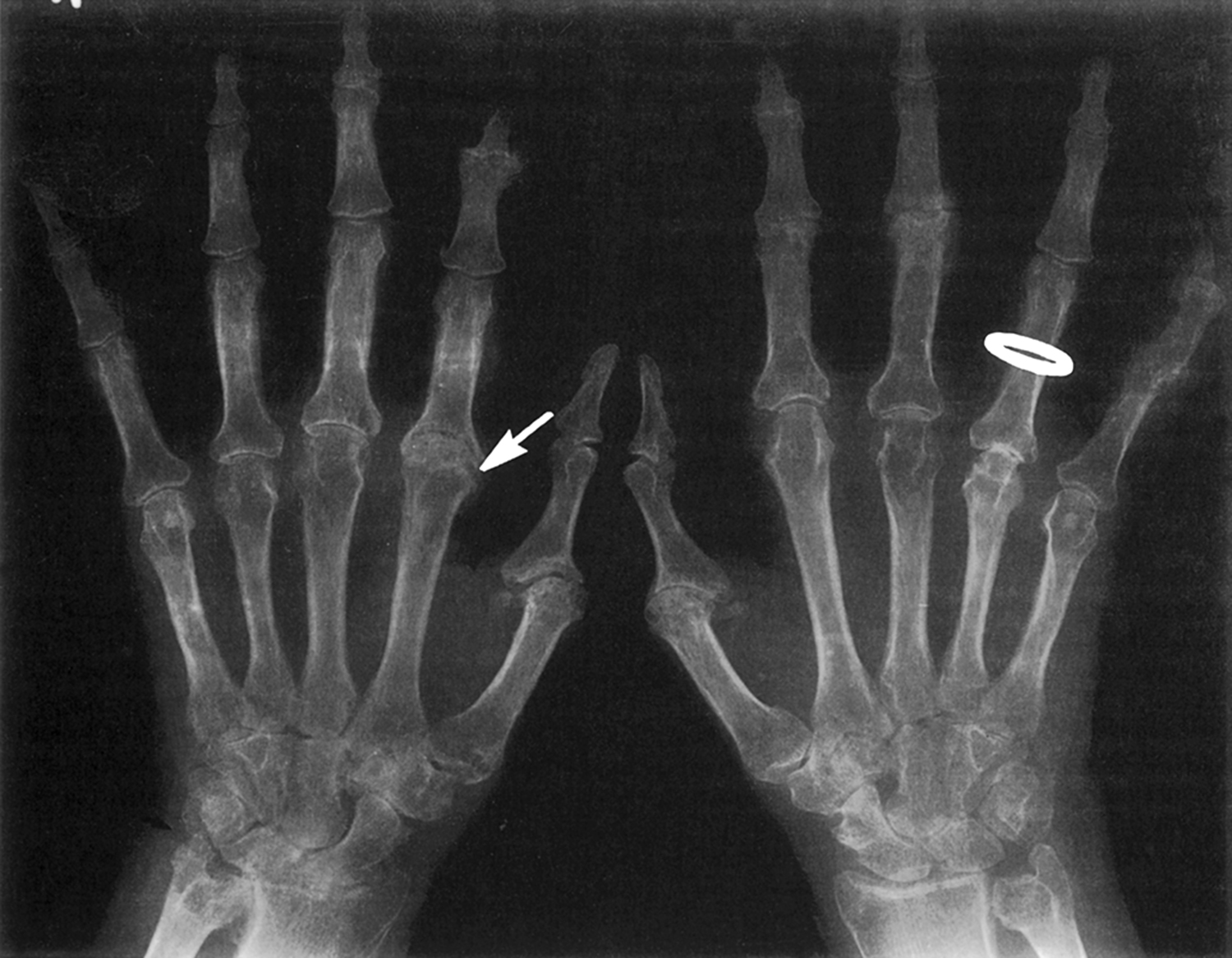
The presence of articular bone erosions is considered the radiographic hallmark of RA. The detection of these bone changes has considerable clinical utility in the diagnosis and monitoring of RA. Different serologies in RA patients are associated with different disease outcomes. In a recent registry study, patients with anti–cyclic citrullinated peptide (CCP) positivity were nearly three times more likely to develop joint erosions . Studies employing the radiographic analysis of progressive changes in articular and periarticular bone have helped to establish that, in general, patients with more extensive bone erosions exhibit more severe disease manifestations and demonstrate worse clinical outcomes . Additional interest in the localized juxta-articular bone changes in RA patients has been generated by clinical trial results showing certain treatment regimens can retard or even prevent the development and progression of focal bone loss . These data indicate the utility of assessment of bone changes as an end point for evaluating treatment response and efficacy.
Much of our present understanding of the pathogenesis of focal bone erosions in RA has come from histopathologic examination of joint tissues and MRI imaging of hands and wrists from patients with RA. The early work of Bromley and Woolley has been particularly informative in providing insights into the cellular mechanisms responsible for the pathogenesis of focal bone erosions . They noted the presence of multinucleated cells with phenotypic features of osteoclasts in resorption lacunae at the pannus–bone interface. Gravallese et al. used in situ hybridization techniques to demonstrate that these multinucleated cells express the full repertoire of phenotypic markers of fully differentiated osteoclasts, including the expression of tartrate-resistant acid phosphatase, cathepsin K, and calcitonin receptor messenger ribonucleic acid (mRNA) . Similar cells frequently line the endosteal surface of the subchondral bone, leading to focal bone erosion. This erosion, combined with inflammatory cell penetration into the deep zones of the articular cartilage, contributes to loss of articular cartilage and joint space narrowing . The marrow adjacent to the sites of subchondral bone erosions is typically replaced by a fibrovascular stroma that is heavily infiltrated with inflammatory cells and adipocytes. Inflammation within the bone marrow and synovium detected by MRI is highly predictive of development of a marginal bone erosion observed by radiograph .
Studies in which inflammatory arthritis was induced in mice with defects in osteoclastogenesis have clearly established that osteoclasts are required for the development of focal joint erosion . Keller et al. studied animal models of inflammatory arthritis with joint destruction similar to the clinical disease observed in RA. The study evaluated the juxta-articular and systemic effects of simultaneous osteoclast inhibition with zoledronate (ZLN) and osteoblast stimulation with parathyroid hormone (PTH). Arthritis-induced mice were randomized to ZLN, PTH, PTH+ZLN, and untreated groups. Locally, they reported no differences in arthritis score, and none of the treatments inhibited bone erosions or stimulated bone formation in the paw. Systemically, all treatments improved bone mineral density (BMD), strength of the femoral neck and mid-diaphysis, and microcomputed tomography parameters of both cortical and trabecular bone . In addition, there was an additive effect of the combination treatment compared with single treatments for most trabecular parameters, including BMD and bone volume fraction.
These findings are supported by studies in human subjects with RA in which treatment targeting osteoclast-mediated bone resorption has been shown to reduce, stabilize, and/or prevent the development of joint erosions . These treatments include denosumab and bisphosphonates. One study compared alendronate and denosumab treatment effects on erosion repair evaluated by high-resolution peripheral quantitative tomography. After 6 months, the width, depth, and volume of the erosions were significantly decreased in the denosumab group, whereas these parameters significantly increased in the alendronate group . Teriparatide, recombinant human PTH (1–34) was investigated as a possible erosion altering medication as well. However, after 1 year of treatment, teriparatide did not reduce erosion volume in the hands or wrists of RA patients compared to the placebo .
Insights into the unique capacity of the rheumatoid pannus to induce osteoclast formation and osteoclast-mediated bone resorption have come from the analysis of RA synovium for the presence of cytokines and other products implicated in the regulation of osteoclast differentiation and activity ( Fig. 53.3 ). These studies have shown that RA synovial tissues produce abundant proosteoclastogenic cytokines, including interleukin (IL)-1, IL-6, IL-11, IL-15, IL-17, IL-23, macrophage colony-stimulating factor, tumor necrosis factor-α (TNF-α), prostaglandins, and PTH-related protein . Results from clinical trials that have targeted IL-1, IL-6, and TNF-α have provided the most compelling evidence that these cytokines play a critical role in the pathogenesis of focal bone erosions . Further evidence is provided from animal models of arthritis using gene transfer or transgenic mouse models. For example, overexpression of TNF-α in normal joints results in pannus formation that leads to focal bone and cartilage destruction . Of interest, IL-1, IL-6, and TNF blockade ameliorate the arthritis in the collagen-induced arthritis model and in humans .
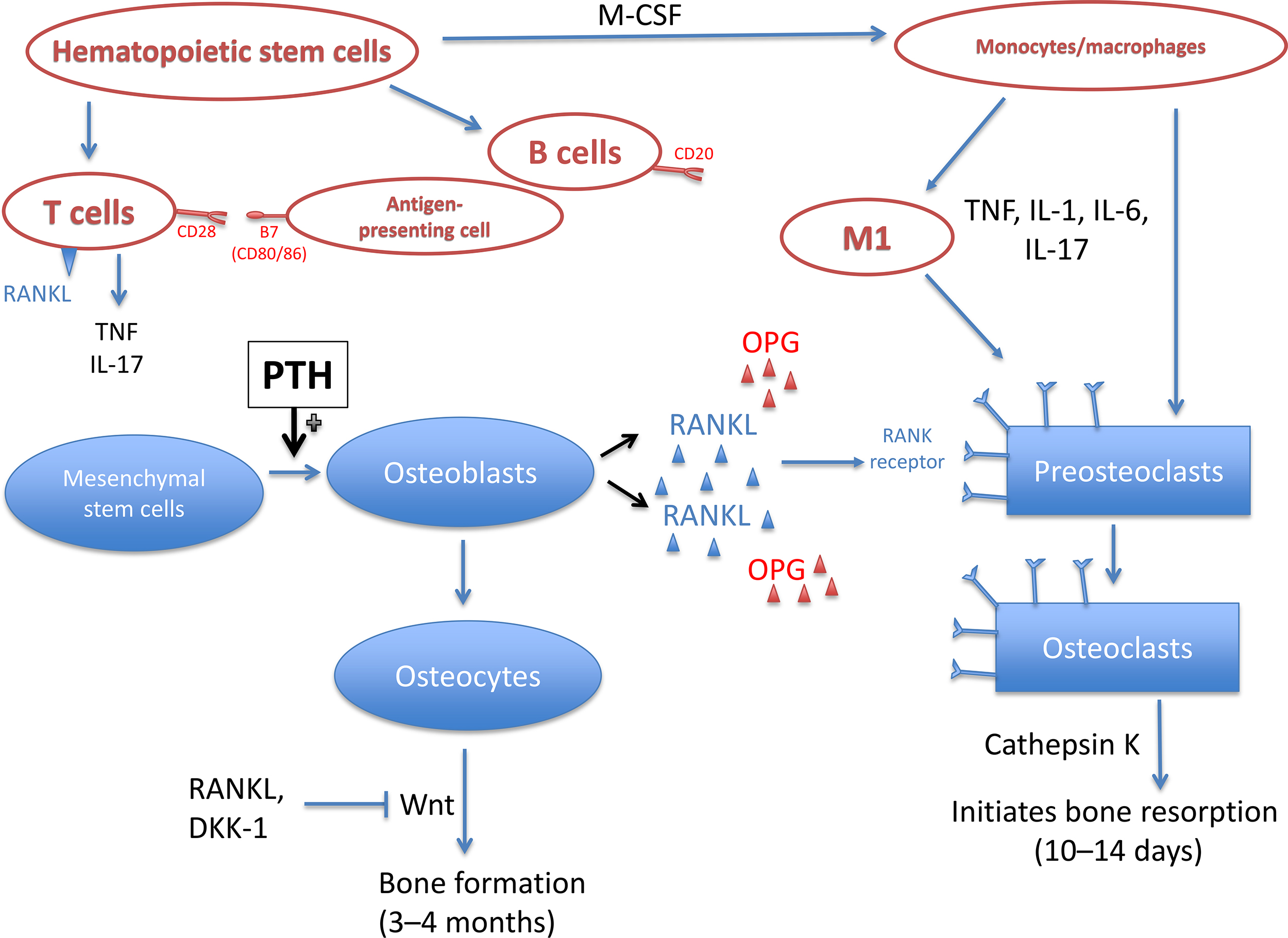
T cells in the synovium produce IL-1, IL-6, and IL-17, which stimulate fibroblast-like synoviocytes to produce receptor activator of nuclear factor kappa-B ligand (RANKL), the potent and essential regulator of osteoclastogenesis . It is a member of the TNF-ligand family of cytokines, and binding to its receptor stimulates several signaling pathways for osteoclast differentiation and activation . The production of RANKL from T cells, synovial fibroblasts, and B cells in RA results in high concentrations of RANKL localized to areas of synovial interaction with bone where bone resorption occurs .
Although all the known inflammatory cytokines can activate RANKL, the activation is through different pathways. In addition to induction of RANKL expression, TNF and IL-6 can also activate osteoclasts in a RANKL-independent pathway. It appears as though the same gene expression characteristics are employed to induce osteoclast formation from precursors as is used with RANKL-induced differentiation, but the TNF/IL-6 pathway is not inhibited by osteoprotegerin (OPG) . TNF-α also amplifies the effects on RANKL on osteoclastogenesis . However, treatment with adalimumab, a TNF-α monoclonal antibody, resulted in improvement in bone erosions for only 6% of patients, suggesting residual inflammation continues to damage the joint structures . IL-6 can induce RANKL expression; however, it can also disrupt downstream signaling pathways for RANK and inhibit precursor differentiation into osteoclasts. Medications or biologic treatments that inhibit IL-6 activity can decrease the number of osteoclasts and inhibit joint damage. IL-1 is produced by various cell types in the joint, including synovial macrophages, fibroblasts, and chondrocytes. The increased levels in the synovial fluid recruit neutrophils and increase blood flow, which results in increased release of matrix metalloproteinases, proteins that induce connective tissue turnover . IL-17, produced by a variety of inflammatory cells, induces the production of inflammatory mediators. Independently, IL-17 induces the production of prostaglandins, nitric oxide, IL-1, and TNF. Prostaglandin E2 is released by osteoblasts in response to IL-17 and contributes to osteoclastogenesis. Although blocking IL-17 in murine models of RA resulted in promising findings of decreased inflammation and articular bone erosion, human trials of RA have not found this to remain true . However, clinical studies studying inhibition of IL-17 in subjects with ankylosing spondylitis (AS) showed a reduction in joint damage in both the axial and appendicular skeleton.
Macrophage polarization may play a role in RANKL overproduction in inflammatory arthritis. Macrophage polarization is the differentiation of macrophages into either M1 or M2 macrophage phenotype depending on the cytokines and transcription factors within the bone/joint microenvironment in which the cell resides. Macrophages in the M1 phenotype are inflammatory and produce proinflammatory cytokines, including TNF-α, IL-1, IL-6, and IL-12, which stimulate other cells to generate RANKL. Conversely, M2 macrophages produce IL-10 and IL-4, which are antiinflammatory cytokines and reduce inflammation. However, when M2 macrophages are exposed to proinflammatory cytokines, the M2 macrophage can switch to M1. The degree to which CD68+, a protein receptor highly expressed by monocytes and tissue macrophages, accumulates in the synovium of the joint has been associated with local joint destruction . Although well-designed preclinical studies are not currently available, M1 cells appear to drive inflammatory bone damage, while M2 cells may assist in bone regeneration. Data for M1 cells’ role in bone resorption can be found in studies evaluating alveolar bone changes in tooth movement. Anti-TNF therapies combined with tooth movement resulted in fewer M1 cells and reduced bone resorption and tooth movement. The effects of M2 cells on bone healing were evaluated in a rodent fracture model and found a negative coupling of decreased TNF and increased M2 levels following bone damage from the fracture decreased inflammation and supported bone healing. In addition, levels of IL-17, a known proinflammatory molecule, is increased in RA and is directly correlated with the ratio of M1:M2 cells in the mucosal tissue of nonhealing areas in bisphosphonate-related osteonecrosis of the jaw .
Bone morphogenetic proteins (BMPs) are known to stimulate bone formation. An in vitro study of macrophages was performed. When they were not exposed to inflammatory signals, the macrophages produced BMPs; however, in the presence of TNF, macrophages lost their ability to produce BMPs . Future studies are needed to clarify the role of macrophage polarization specifically in rheumatic diseases, but this preliminary data indicates that it may contribute to the erosions from local inflammation seen in RA.
RANKL was originally cloned and characterized as a product of activated T cells and was designated as TNF-related activation-induced cytokine. T cells within the RA synovium are a primary source of RANKL, as are synovial fibroblasts . Direct evidence that RANKL has an important role in the pathogenesis of focal bone erosions in inflammatory arthritis is provided by the studies of Kong et al. . Lewis rats were injected with Mycobacterium tuberculosis to induce arthritis, then they were injected with OPG at the onset of inflammation, and monitored via hindpaw water displacement for inflammation severity. While the increase in RANKL was not balanced by an increase in its inhibitor, OPG, in the joints, Kong et al. showed treatment with OPG almost completely blocked the development of cortical and trabecular bone loss in a rat model of adjuvant arthritis with minimal effects on joint inflammation or pannus formation. Pettit et al. later demonstrated that bone resorption could be separated from inflammation, reporting that RANKL knockout mice had a reduction in bone erosion without a reduction in synovial inflammation . However, bone formation seems to be more closely tied to a reduction in joint inflammation. Inflammation limits the differentiation of osteoblast precursor cells, and therefore any potential healing of joint erosions gets affected. Resolution of inflammation allowed for differentiation of the precursor cells, and partial repair of existing erosions .
Although the optimal therapy for RA should reduce or abolish inflammatory synovitis, therapies that reduce the damage to bone or articular cartilage in the absence of effects on the synovitis may have a potentially important role in treatment. Agents that specifically target osteoclastic bone resorption have been evaluated in several clinical studies in RA patients and in animal models of arthritis. In general, studies employing bisphosphonates have shown a beneficial effect on systemic bone loss. However, with the exception of a more recent study by Jarrett et al., bisphosphonates have not generally been effective in arresting the development of focal bone erosions . In contrast, studies in animal models of arthritis have shown bisphosphonates do retard, or even prevent, focal bone loss . Although the validity of these models as surrogates for RA can be challenged, they provide a convincing argument for additional trials with antiresorptive therapies in RA. In more recent studies, blockade of RANKL with a human monoclonal antibody was shown to reduce the development and progression of joint erosions and protect from systemic bone loss, supporting the concept that osteoclasts mediate the focal resorptive process and identifying RANKL as a key regulator of osteoclast formation and activity .
An interesting study recently reported how the inflammatory mediators from the synovitis within the joint can be transported to the juxta-articular bone. Studying both cadaver knees and an inflammatory mouse model, investigators performed 3 T MRI and histology and found vascular channels penetrating cortical bone in knees of nonarthritic mice adjacent to the cruciate ligaments. Histology confirmed the location of vascular channels was associated with subclinical changes, including subchondral bone damage and microcyst formation. In the inflammatory mouse model, these channels provided a site for inflammatory mediator entry into the juxta-articular bone, resulting in osteoclast activation and bone marrow inflammation shown on MRI. These observed vascular channels adjacent to the ligament entheses are common sites of subtle bone marrow pathology in nonarthritic joints and appear to augment bone damage in inflammation .
Overall, the interactions between the immune system and the skeletal system in patients with RA result in increased osteoclastogenesis with reduced osteogenesis, ultimately resulting in local bone loss. It is also possible that the inflammatory mediators that cause juxta-articular bone loss may be transported by vascular channels that connect the synovial-lined joint with the subchondral bone adjacent to the joint.
53.4
Generalized bone loss
Several studies have provided evidence for generalized bone loss in the axial spine and appendicular skeleton distant from synovial-lined joints in patients with RA . Identification of the independent role of the systemic inflammatory process in bone remodeling has been difficult to establish due to a number of factors, including reduced mobility, variation in the level of disease activity and duration, and the concomitant use of glucocorticoids or other immunosuppressive therapies that can influence both appendicular and axial bone mass . In addition, many of the epidemiological studies have been flawed by weak study designs, such as small numbers of subjects and inconsistent methods of measurement of physical activity, including reliance on study subject self-report, which reduced the ability to determine the effects of RA, and the medications, on overall bone mass. Despite these limitations, there does appear to be an increase in the risk for fracture among patients with RA, most recently supported by a systematic review and metaanalysis containing 25 studies . Pooled incidence rates for all fractures from these studies were 33 per 1000 person-years with 15.31 for fragility fractures, and pooled relative risk for three studies of cohorts with control groups was 1.52 (95% CI 1.07–2.14) . Two of the reviewed studies included only women, and the relative risk of fracture at all sites was 1.77 (95% CI 1.19–2.64). When these studies were evaluated together, the independent predictors of fracture risk in RA subjects were elevated C-reactive protein (CRP) levels, reduced mobility and ability to perform activities of daily living, longer disease duration, and higher disease activity using the disease activity score 28.
Several studies have suggested that in patients with RA there is a relationship between systemic osteoporosis and disease activity . For example, in the study by Hooyman et al., regression analysis suggested that in addition to duration of disease and disability level, age and glucocorticoid exposure were the major risk factors for fractures . Similar findings were obtained by Michel et al., who analyzed data from five Arthritis, Rheumatism and Aging Medical Information System centers . Using multivariate analysis, they identified an association between fracture risk and the years taking prednisone, disability, age, lack of physical activity, female sex, disease duration, impaired grip strength, and low body mass. More recently, BMD was assessed in 394 RA patients from the Oslo County Rheumatoid Arthritis Registry . The authors reported a twofold increase in osteoporosis (defined as a T score of 2.5 standard deviations or more below the mean) in these female RA patients (20–70 years) compared to the BMD of healthy women ages 20–69 years. A linear regression model was used to determine individual predictors of BMD. In the final model, older age, low body weight, current use of glucocorticoids, and lower functional status (assessed by modified Health Assessment Questionnaire score) were significant predictors of reduced bone mass.
A recently discovered predictor of BMD in RA patients is the serum levels of anti-CCP. Multivariable sensitivity analysis of 61 RA patients with elevated anti-CCP antibodies noted that a high-serum anti-CCP of greater than 60 U had a negative association with femoral neck BMD [beta coefficient of −0.054 (95% CI −0.097 to −0.010) with a P value equal to .016].
In addition, lean mass index was positively associated with femoral neck BMD ( P =.018), while sex, age, disease duration, and knee flexion strength were not . Citrullination is a normal physiological process; however, it occurs inside of dying cells, protecting the immune system from contact with the citrullinated proteins. In situations of massive cell death, as can be seen with inflammation, pepdidylarginine deiminase enzymes and citrullinated proteins leak from dying cells and come into direct contact with the immune system. It is not clear why some individuals develop antibodies to citrullinated proteins and others do not, but it is thought to be partially dependent on the MHC alleles of the affected individuals .
Although epidemiological studies indicate that glucocorticoid use in patients with RA is a risk factor for systemic osteoporosis, the effects of glucocorticoids on bone mass changes have been difficult to determine. In part, this is related to the capacity of low doses of glucocorticoids to reduce inflammation and improve symptoms and functional capacity in patients with RA. In support of this concept, in cross-sectional and longitudinal studies by Sambrook, patients with RA had lower levels of BMD than did normal controls, but no difference was noted in the progression of bone loss in RA patients treated with or without low-dose glucocorticoids . Lane et al. examined the association of glucocorticoid use with BMD in a community-based sample of ambulatory Caucasian women aged 65 and over with or without RA . They found that women with RA who were current users of glucocorticoids had the lowest bone density at both the appendicular sites and at the hip. Women who had never used glucocorticoids also had a reduced bone density compared to the control population. They concluded that the lower appendicular and axial bone mass observed in women with RA is not completely attributable to the use of glucocorticoids. The lower BMD in the women on glucocorticoids could also be accounted for by their lower functional status and a low-grade chronic inflammatory state. Other studies have suggested that even low-dose glucocorticoids may have a detrimental effect on bone density. For example, in a cross-sectional analysis employing quantitative computed tomography, Laan et al. demonstrated that low doses of glucocorticoids (mean dose, 6.8 mg prednisone/day) were associated with reduced bone density compared to nonglucocorticoid-treated patients . In a case–control analysis of 112 RA patients by Saag et al., long-term low-dose glucocorticoid was a significant predictor of adverse events, including fractures, gastrointestinal events, and infections .
There are conflicting results with respect to the mechanisms responsible for the reduced bone mass in patients with RA related to bone turnover markers and other systemic markers of risk of bone turnover. In a longitudinal study, Gough et al. analyzed biochemical markers of bone turnover in a series of 232 patients with RA . Over a 2-year period they detected a significantly greater rate of bone loss in the RA patients compared to the controls (greater than 3% at the spine and 5% at the hip). Results showed elevated urinary pyridinoline and deoxypyridinoline, indicating that there were increased levels of biochemical markers of bone resorption in patients with RA. Of interest, the levels of these markers were highly correlated with CRP levels. The increase in bone resorption was not accompanied by changes in bone formation markers, as assessed by serum alkaline phosphatase activity and procollagen I C-terminal propeptide concentrations. Similar observations have been made in other studies, which reported increases in urinary markers of bone resorption were associated with active disease and rapid changes in BMD . The findings from these studies must be interpreted with caution, since in some instances the patients were receiving glucocorticoids. In contrast to the results from studies in which bone markers have been used to assess bone remodeling, the histomorphometric analysis of bone biopsies from patients with RA indicates that the cellular basis for the reduced bone mass is related to a decrease in bone formation rather than an increase in bone resorption . These discrepancies could reflect differences in the stages of the disease in which bone remodeling was being evaluated, the confounding effects of glucocorticoid treatment and disease activity in the patient populations, and the site of which the bone biopsy was obtained, for example, the iliac crest, is not near a joint with synovitis from RA.
Although attention has focused on the periarticular and focal bone destruction in RA, an additional finding is the uncoupling of bone resorption and formation with virtual absence of new bone formation and repair. The studies by Diarra et al. demonstrated that the defect in bone formation can be partially attributed to increased dickkopf-1 (DKK-1), a potent inhibitor of the wingless (Wnt) signaling pathway, which plays an important role in osteoblast-mediated bone formation . They showed that synovial fibroblasts, endothelial cells, and chondrocytes were the major sources of DKK-1 , and that TNF-α was a potent inducer of DKK-1 in RA synovial fibroblasts, implicating this proinflammatory cytokine in both bone formation and bone resorption. These observations have been confirmed by Walsh et al., who showed that RA synovium produces additional Wnt family antagonists, including members of the DKK and secreted Frizzled-related protein families . The correlation was once again reported by Ruaro et al. when they evaluated trabecular bone score in RA and systemic sclerosis with DKK-1 levels . In the Diarra studies, they also showed that inhibition of DKK-1 with a blocking antibody not only corrected the defect in bone formation but surprisingly also suppressed osteoclast-mediated bone resorption. Finally, levels of DKK-1 were found to correlate with degree of bone erosion, decrease in BMD, and disease activity . The effect on osteoclast-mediated bone resorption was attributable to downregulation of RANKL and upregulation of OPG in the synovial tissue. These observations have clear implications with respect to future therapeutic strategies targeting Wnt pathway antagonists to prevent pathologic bone loss in RA .
As previously mentioned, denosumab’s mechanism of action is osteoclast inhibition. This inhibition was found to result in a skeleton wide decrease in bone turnover and increase in BMD at both lumbar spine and total hip over a 12-month period . Bisphosphonates have been shown to improve BMD; but in patients with well-controlled disease, BMD was only significantly improved at the total spine, not whole body or lower limb . In another study, bisphosphonates and denosumab were both shown to significantly improve lumbar and total hip BMD after 24 months of treatment, with denosumab treatment resulting in improvement as early as 6 months . A 10-year follow-up study for RA patients on TNF-ɑ inhibitors or nonbiologic DMARDs found both treatments to be equally effective at slowing rates of BMD loss .
In summary, inflammatory cytokine stimulation of RANKL and DKK-1 results in altered Wnt pathway activity in the osteoblast, leading to reduced synthesis of OPG, and ultimately causing accelerated osteoclast resorption and decreased bone formation.
Inhibition of the Wnt signaling antagonist, sclerostin, can increase bone formation in postmenopausal women with osteoporosis and has been studied in animal model of inflammatory bone loss . Sclerostin inhibition in a collagen-induced murine model of arthritis, resulted in full restoration of BMD to control levels at all skeletal sites evaluated; however, sclerostin inhibition did not change the local inflammatory-induced bone erosions . If the sclerostin antibody is approved for the treatment of postmenopausal osteoporosis, then it can potentially be tested in RA subjects to improve bone mass.
53.5
Juvenile idiopathic arthritis
JIA is a systemic inflammatory joint disorder characterized by chronic synovitis affecting diarthrodial joints. While estimates for the prevalence of JIA in the general population vary, a large systematic review based on European populations reports a prevalence of 32.6/100,000 . Similar to the findings in patients with RA, children with JIA show evidence of multiple distinct patterns of bone loss, including focal marginal erosions, juxta-articular osteopenia, and generalized osteoporosis. In some instances, the peripheral joint inflammation is accompanied by enthesopathy and sacroiliitis. The disease may develop at any age during childhood and tends to affect girls more frequently than boys, although the sex ratios vary in different subsets of the disease. Three major patterns or subsets of JIA have been described: pauciarticular, polyarticular, and systemic. It is not clear whether these represent different disease entities that share the ability to produce joint inflammation, or whether they are manifestations of various responses to common pathogenic factors.
There is little information concerning the histopathologic events associated with focal bone erosions and juxta-articular osteopenia in children with JIA. It is likely, however, that the pathological processes responsible for the bone and cartilage destruction are similar to those that described in adults with RA. As in RA, generalized loss of bone mass is a common feature of all of the forms of JIA. The risk factors are similar to those associated with systemic osteoporosis in RA, including the effects of medications, reduced level of physical activity, dietary deficiencies, and the adverse systemic effects of inflammatory mediators and cytokines . The importance of disease activity on bone mass changes in children with JIA is demonstrated by the studies of Reed et al. , who evaluated radial BMD over a 3-year period in children with JIA. Improvement in the disease activity was associated with an increase in BMD, although levels remained below the normal values.
Hopp et al. studied spinal bone density in 20 children with active JIA and found reduced values in postpubertal girls compared with healthy controls . They noted that adolescents with active disease may be particularly vulnerable to the impact of the inflammatory process on the skeleton, in part because of the rapid skeletal acquisition that is normally associated with this stage of development. Of interest, bone mass in prepubertal girls did not differ from controls at any of the skeletal sites examined. Henderson et al. studied total-body bone mineral content in nonglucocorticoid-treated postpubertal females with JIA . They found that approximately 30% of these adolescents/young adults with mild-to-moderate JIA had low bone mass determined by Z -score of the radius. Of interest, using stepwise linear regression they found that the predictor variable that significantly contributed to total-body bone mineral content was lean body mass. Even when evaluating prepubertal adolescence with no exposure to glucocorticoids, polyarticular JIA patients had a significantly reduced distal radius Z -score. Overall, 7/10 studies included in a large metaanalysis found a modest but significant decrease in BMD for JIA patients, while 3/10 studies found no significant difference .
A unique aspect of the skeletal pathology in JIA is the effect of the inflammatory process on skeletal growth. Because the joint disease affects children during the period of skeletal acquisition, there is often dramatic linear growth retardation in addition to the adverse effects on bone remodeling . The suppression of bone formation in children, especially during the pubertal growth period, may have a major adverse effect on the achievement of optimal peak bone mass. The failure to achieve optimal peak bone mass predisposes these individuals to an increased fracture risk in adulthood . Fracture risk for JIA children as adolescence may be slightly increased, but data is conflicting. In a population-based cohort study, there was no difference in fracture rate between patients and controls over a 2-year period. However, in a larger population database study, 6.7% of patients had fractures compared to 3.3% of controls ( P <.001) .
Pereira et al. analyzed biochemical markers of bone remodeling in children with JIA, and observed a low bone formation rate in younger children, and increased bone resorption in the older age-group, over 12 for girls and 14 for boys . Similar results have been reported by Pepmueller et al., who measured BMD and markers of bone remodeling in 41 children with JIA. Bone density was reduced at all sites. Low levels of osteocalcin and bone alkaline phosphatase were consistent with reduced bone formation . They noted that laboratory markers of disease activity were highly correlated with decreases in markers of bone formation, but not with those of bone resorption. In general, the findings were similar in children with pauciarticular and polyarticular JIA, although the reduction in bone mass was greatest in the children with the polyarticular form of arthritis.
53.6
Glucocorticoids in children
As in adults, glucocorticoids are a mainstay of treatment for adolescent rheumatic diseases, but these come with multiple unwanted side effects. When looking at 15 children with rheumatic disease treated with glucocorticoids for an average of 6.2 years with recent vertebral fractures compared to age-matched controls, treated children had a significant decrease in trabecular thickness, osteoid thickness, osteoblast surface, and increased trabecular separation. However, the glucocorticoid-treated children also had a hypermineralized profile, which is correlated to the dose of glucocorticoids . In the Steroid-Induced Osteoporosis in the Pediatric Population study, which included 50 JIA patients, patients with systemic and nonsystemic disease had significantly decreased BMD measured at the lumbar spine . Yang et al. studied wild-type mice and cathepsin K knockout mice and examined the cell populations of the primary and secondary spongiosa before and after glucocorticoid treatment. Cathepsin K was chosen because genetic loss of cathepsin K inhibits osteoclastic bone resorptive activity and results in a high bone mass mouse phenotype, similar to osteopetrosis. When treated with prednisolone, cathepsin K knockout mice maintained their trabecular bone volume, while wild-type mice had decreased trabecular bone volume. In addition, wild-type mice treated with glucocorticoids had fewer osteoclasts, tartrate-resistant acid phosphatase, PDGF-type BB copositive cells, type H endothelial cells, and osteoblasts compared to untreated wild-type mice, and increases in all four cell types listed previously in the cathepsin K knockout mice treated with glucocorticoids. When wild-type mice treated with glucocorticoids received a cathepsin K inhibitor, there was no change in quantity of these cells. These data suggest that cathepsin K, or other osteoclast inhibitors, may prevent glucocorticoid-induced osteoporosis in adolescence with active bone growth .
53.7
Osteoporosis treatment in children
There are currently no guidelines specifying treatment for osteoporosis in children. Patients that may benefit from treatment are those with Z -scores <−1.5, history of fracture, or patients receiving glucocorticoid treatment for >6 months .
53.8
Seronegative spondyloarthropathies
The seronegative spondyloarthropathies represent a heterogeneous group of inflammatory disorders that include AS, reactive arthritis, psoriatic arthritis (PsA), arthritis associated with inflammatory bowel disease, and juvenile-onset spondyloarthropathy. In the North American population, the prevalence of AS has been reported at 31.9/10,000 , while psoriatric arthritis in the US population has a prevalence of 0.06%–0.25% . Although these disorders may produce inflammation of peripheral joints, inflammation of the entheses (sites of tendinous or ligamentous attachment to bone), especially in the axial spine, represents the pathological hallmark of the spondyloarthropathies. Synovial hyperplasia, lymphoid infiltration, and pannus formation are frequently observed in affected joints. However, the inflammation is usually restricted to a limited number of joints and the pattern of distribution is typically asymmetrical, affecting distal as well as proximal joints.
Insights into the topographical localization of the inflammatory joint pathology has been provided by the introduction of MRI techniques that are capable of delineating sites of bone and connective tissue inflammation in patients with spondyloarthritis (SpA) . With these techniques, it has been possible to confirm that the inflammatory process that accounts for the initial joint pathology frequently begins at the enthesis. These changes are visualized as focal soft tissue edema that is maximal at regions adjacent to the entheseal insertions in peripheral joints .
There have been relatively few studies of the histopathology associated with the entheseal and synovial inflammation in the spondyloarthropathies. One study examined cadavers to evaluate the association between proximity to entheses’ inflammation and degree of damage and noted that histopathology of synovial tissue showed more synovial villus formation near areas of enthesis inflammation . In addition to the differential localization of the inflammation to include the enthesis, the synovial inflammatory process in the spondyloarthropathies, unlike the joint inflammation in RA, may be accompanied by evidence of increased bone formation. Braun et al. used computer-assisted tomography to obtain biopsies of the sacroiliac joint in a series of patients with AS . Immunohistologic examination of the tissue revealed dense infiltrates of T lymphocytes (CD4+ and CD8+) and macrophages (CD14+) in the synovial lining accompanied by localized nodules containing active foci of endochondral ossification. In situ hybridization demonstrated a high signal for TNF-α in the inflammatory cells. Of interest, abundant transforming growth factor (TGF)-β2 mRNA was expressed in cells at sites of new bone formation. These authors suggested that local production of bone growth factors, such as TGF-β, by the inflammatory cells within the synovium could be responsible for the new bone formation. This process could account for bony ankylosis of the sacroiliac joints that is characteristic of AS and other forms of seronegative spondyloarthropathy. Lories et al. studies involving the DBA/1 mice that spontaneously develop an inflammatory arthritis that recapitulates the excessive bone formation characteristic of AS showed that delivery of noggin, a BMP antagonist, attenuated periarticular new bone formation . They also examined the entheseal biopsies from patients with AS and identified phosphorylated SMAD 1/5 expression in the synovial tissues. These findings are consistent with local activation of the BMP signaling pathway and provide further evidence that BMPs are involved in the new bone formation associated with the spondylitis. Further evidence supporting this role for bone growth factors is provided by the characterization of transgenic animals overexpressing BMP-6 . These animals develop psoriatic skin lesions and an osteoarthropathy similar to the joint and spine pathology of psoriasis.
Alterations in IL-23 expression can begin to explain the skeletal pathologies in SpA. The HLA-B27 allele related to many seronegative spondyloarthropathies induces IL-23 production, resulting in osteoclastogenesis and bone resorption systemically due to an upregulation of inflammation and BMP-7 . Overexpression of IL-23 has also been shown to promote enthesial inflammation and enthesial bone formation .
BMP-7 is another relevant BMP in the development of AS, RA, and PsA. Serum levels of BMP-7 were shown in one study to be significantly elevated over levels found in healthy controls, and these elevations were linked to elevated disease activity scores. The skeletal pathology of PsA is quite different than that of RA. While in RA there is a lack of osteogenesis and juxta-articular osteopenia, studies show unchanged periarticular bone density in PsA hand radiographs, and vigorous new bone formation in a pathologic manner, resulting in bony nodules both localized and distant from sites of resorption . However, PsA patients have lower axial BMD, supporting an aspect of systemic bone loss from a chronic inflammatory state in this rheumatic disease as well. Although some studies report an increased risk of fracture in PsA patients, others have found no association. To date, it appears as though the combination of both localized and systemic bone formation, over a period of years in these subjects may have both systemic bone loss and increased fracture risk.
In addition to the previously mentioned potential role of the BMP/TGF-β pathways in contributing to the periarticular bone formation in the spondyloarthropathies, there is evidence that the inflamed synovium and entheseal tissues may be deficient in inhibitors of the signal pathways regulating bone formation . As described in the preceding section, inflamed synovial tissues are an abundant source of several potent inhibitors of the Wnt pathway (e.g., DKKs), which regulates osteoblast-mediated bone formation . In contrast to the relationship between RA and DKK-1, patients with AS do not exhibit elevated DKK-1 levels, providing a potential link between the enhanced articular bone formation and the low levels of this inhibitor of bone formation . In fact, additional studies reported an inverse correlation between DKK-1 serum levels and syndesmophyte development . Although another study showed elevated DKK-1 in AS patients, they also found that it was a dysfunctional form of the protein . These findings seem to explain the low-serum DKK-1 levels and the osteophyte formation observed in PsA patients. A protein with similar effects in both RA and SpA is IL-17. This proinflammatory and osteoclastogenic cytokine is increased in the serum of AS and PsA patients and appears to correlate with disease activity .
A more recently discovered protein associated with bone metabolism in AS and PsA is prostaglandin E2. In mice, cyclooxygenase-1 and cyclooxygenase-2 (COX2) enzymes are associated with impaired osteoblast maturation and delayed bone fracture healing. In clinical studies of AS/PsA patients, this translates into inhibition of syndesmophyte formation in the spine. Continuous treatment of AS/PsA patients with these inhibitors (nonsteroidal antiinflammatory drugs (NSAIDs) or COX2 inhibitors) with and without TNF blockers has shown reduction in syndesmophyte formation in humans . Additional studies evaluating high-dose or continuous NSAID use compared to intermittent or low-dose NSAID use found that continuous or high-dose therapy significantly reduced modified Stoke Ankylosing Spondylitis Spine Score .
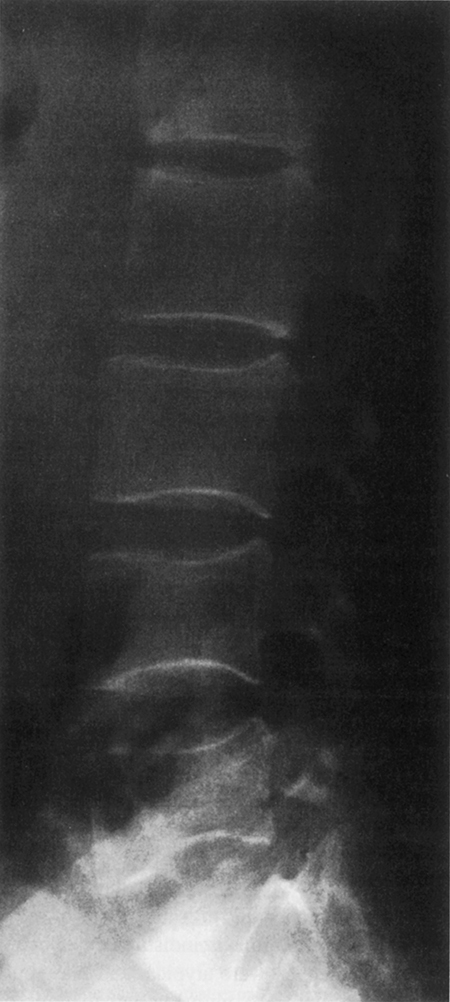
Despite the tendency of patients with spondylitis to develop bony ankylosis of the spine, there is evidence of vertebral osteopenia and an increased incidence of fractures ( Fig. 53.4 ). Will et al. evaluated a series of patients with early AS using dual-photon absorptiometry and observed a significant reduction in BMD in the lumbosacral spine and hip early in disease before bony ankylosis and spinal immobility developed . They speculated that the reduced bone mass was related to the adverse effects of inflammation on bone remodeling. Ralston et al. prospectively evaluated a group of 111 patients with AS , of which 15 developed radiographic evidence of vertebral compression fractures. These patients tended to have a greater degree of spinal deformity and less spinal mobility than individuals without fractures. He concluded that vertebral compression fractures secondary to spinal osteoporosis were a common but frequently unrecognized complication of AS, and that they contributed to the pathogenesis of spinal deformity and back pain. Of interest, BMD of the appendicular skeleton was normal, suggesting that osteoporosis in these patients is primarily localized to the axial spine. Similar observations have been reported by Devogelaer et al., who observed that males with AS tended to develop significant bone loss in the vertebral bodies but exhibited bone density comparable to the controls in the appendicular skeleton . BMD can be improved by treatment with TNF-α blockers, but other DMARDs, such as sulfasalazine and methotrexate, have not been studied for their effects on BMD in AS . The effect of biologic treatments on fracture risk in AS needs to be evaluated.