54.1
Introduction
Since the kidney is essential for regulation of mineral metabolism, bone disease is an inevitable consequence of renal damage. Treatment is challenging due to the complex physiology and the dearth of clinical data and is complicated by concern about vascular calcifications. The term CKD-MBD (chronic kidney disease–mineral and bone disorder) highlights the connections between the biochemical and hormonal abnormalities of mineral metabolism, the vascular calcifications, and the skeletal disease .
54.2
Grades of chronic kidney disease
The grades of CKD are defined by the presence of a reduced glomerular filtration rate (GFR), or by other indicators of renal disease, particularly the underlying cause and degree of albuminuria. In clinical practice the GFR is estimated (eGFR) using a formula based on the serum creatinine, age, race, and gender of a person, expressed as GFR per body surface area, with grades shown in Table 54.1 . CKD grades 1 and 2 refer to kidney diseases associated with high/normal or mildly decreased GFR, for example, early diabetes or conditions interfering with tubular transport, such as Fanconi’s syndrome. These patients may have bone disease, due to acidosis or hypophosphatemia, but that is beyond the scope of this chapter. Here, we will consider the management of patients with CKD G3–G5. Grade 3A is defined as eGFR between 59 and 45 mL/min/1.73 m 2 ; G3B between 44 and 30; G4 between 39 and 15; G5 less than 15 and G5D receiving dialysis. Findings of CKD-MBD (abnormal mineral levels or related hormones, bone disease, or vascular calcifications) first typically manifest in G3B to G4.
| Grade | eGFR mL/min/1.73 m 2 | eGFR categories | Albuminuria categories | ||
|---|---|---|---|---|---|
| A1 | A2 | A3 | |||
| Normal | Moderately increased | Severely increased | |||
| G1 | ≥90 | Normal or high | 55.6 | 1.9 | 0.4 |
| G2 | 60–89 | Mildly decreased | 32.9 | 2.2 | 0.3 |
| G3a | 45–59 | Moderately decreased | 3.6 | 0.8 | 0.2 |
| G3b | 30–44 | Moderate to severely decreased | 1.0 | 0.4 | 0.2 |
| G4 | 15–29 | Severely decreased | 0.2 | 0.1 | 0.1 |
| G5 | <15 | Kidney failure | <0.1 | <0.1 | <0.1 |
A systematic review of studies worldwide found the median prevalence of CKD in adults is 7.2%, and in those older than 64 this increases to about 30% . Women had a higher rate than men, African-Americans had a lower prevalence than Caucasians, and Asian populations had a relatively high prevalence. The prevalence will vary depending on the method used to calculate kidney function . The methods which use serum creatinine depend on the muscle mass. Cystatin C, another marker used to determine GFR, is independent of muscle mass and is a more sensitive method for detecting CKD, particularly in elderly patients or those with muscle disorders.
54.3
Epidemiology
54.3.1
Fracture rates
Abnormal bone strength resulting from CKD can lead to increased fragility and an increased risk of fracture. Pendras et al. reported their experience with the first 22 patients to receive long-term hemodialysis, for whom bone and mineral disorders emerged as one of the most troublesome complications, with fractures occurring in 47%. Since then, reported prevalence rates have varied from 4% to 44% ( Table 54.2 ) in dialysis populations . An international study of hemodialysis patients in the Dialysis Outcomes and Practice Patterns Study found regional differences in rates of hospitalized fractures, from 1.2%/year in Japan to 4.5%/year in Belgium .
| Author | Year | N | Patients | Country | Prevalence spine (%) |
|---|---|---|---|---|---|
| Parfitt | 1972 | 16 | HD | United States | 25 |
| Yamaguchi | 1996 | 124 | HD | Japan | 11 |
| Atsumi | 2000 | 187 | HD | Japan | 21 |
| Jamal | 2002 | 104 | HD>55 years | Canada | 33 |
| Urena | 2003 | 70 | HD | France | 7 |
| Inaba | 2005 | 114 | PD>65 years | Japan | 18.4 |
| Elder | 2006 | 242 | Pre-Tx HD, PD | Australia | 28 |
| Mares | 2009 | 72 | HD | Czech Rep. | 21 |
| Iimori | 2012 | 485 | HD | Japan | 6 |
| Fusaro | 2013 | 387 | HD | Italy | 53 |
| Author | Year | N | Patients | Country | Prevalence any (%) |
|---|---|---|---|---|---|
| Pendras | 1966 | 19 | First HD patients | United States | 47 |
| Rubini | 1969 | 29 | HD | United States | 27 |
| Parfitt | 1972 | 16 | HD | United States | 44 |
| Yamaguchi | 1996 | 124 | HD | Japan | 10 |
| Gerakis | 2000 | 62 | HD | Greece | 11 |
| Jamal | 2002 | 104 | HD>55 years | Canada | 52 |
| Urena | 2003 | 70 | HD | France | 30 |
| Negri | 2004 | 65 | PD | Argentina | 9.2 |
| Ersoy | 2006 | 292 | PD | International | 10 |
| Jamal | 2006 | 52 | HD>50 years | Canada | 52 |
| Simunovic | 2015 | 767 | HD | Croatia | 4 |
| Evenepoel | 2018 | 518 | HD pre-Tx | Netherlands | 5.8 |
| Nakanishi | 2018 | 293 | HD | Japan | 14.2 |
Hip fractures seriously impact quality of life and are associated with increased mortality in patients with CKD. The incidence in dialysis patients ranges from 0.6 to 2.2%/year ( Table 54.3 ). The incidence of hip fracture for patients commencing dialysis from 1989 to 1996 in the United States was 4.4-fold that of residents of Olmstead County . Subsequently, hip fracture rates in US dialysis patients older than 67 increased from 1996 until 2004 and then declined, although rates in 2009 remained higher than in 1996 . Utilizing the same United States Renal Data System, another study found that between 2003 and 2011, patients with CKD-G5 over the age of 65 experienced a 15.3% decline in rates of hip fracture, but there was no change in rates for younger patients . Medicare data also showed that hip fracture rates increased steadily from 1993 to 2004 and decreased through 2010 . In Japanese dialysis patients, hip fracture incidence decreased between 2008 and 2013 in women, whereas in the general population the rate increased . A metaanalysis of 14 cohorts found the overall risk of hip fracture in dialysis or transplant patients is five times higher than in the general population .
| Author | Year | N | Patients | Country | Incidence any (%/year) |
|---|---|---|---|---|---|
| Stehman-Breen | 2000 | 5K | HD | United States | 0.69 |
| Coco | 2000 | 1.2K | HD | United States | 1.39 |
| Alem | 2000 | 32K | HD men | United States | 0.74 |
| Women | 1.36 | ||||
| Ball | 2002 | 101K | USRDS on tx list | United States | 0.29 |
| Danese | 2006 | 9K | USRDS | United States | 0.65 |
| Jadoul | 2006 | 12K | HD | International | 0.89 |
| Kaneko | 2006 | 7K | HD | United States | 1 |
| Wakasugi | 2013 | 128K | HD | Japan | 1.13–1.07 |
| Arneson | 2013 | 203K | HD | United States | 1.2 to 2.2 |
| Lin | 2014 | 51K | HD | Taiwan | 0.8 |
| Maruyama | 2014 | 187K | HD | Japan | 1.1 |
| Maravic | 2014 | 29K >50 | CKD5D >50 | France | 1.23 |
| Chen | 2014 | 64K | HD+PD | Taiwan | 1.36 |
| Mathew | 2014 | 929K | HD | United States | 1.0–2.2 |
| Kim | 2016 | 4.8K | CKD and HD | United States | 0.389 |
| Hansen | 2016 | 7.5K | HD | Denmark | 1.89 |
| Author | Year | N | Patients | Country | Incidence any/year |
|---|---|---|---|---|---|
| Piraino | 1988 | 31 | HD | United States | 0.1–0.2 |
| Block | 2004 | 40,538 | HD | United States | 0.52 |
| Jadoul | 2006 | 12,782 | HD | International | 2.56 |
| Mitterbauer | 2007 | 1774 | HD | Austria | 1–3 |
| Iimori | 2012 | 485 | HD | Japan | 1.9 |
| Nair | 2013 | 409K | CKD5D>67 | United States | 2.93 |
| Tentori | 2013 | 34K | HD | International | 1.2–4.5 |
| Wagner | 2014 | 935K | HD | United States | 1.25–2.53 |
| Naylor | 2014 | 24,864 | CKD5 women>65 men >65 | Canada | 3.2 1.6 |
| Hansen | 2016 | 7566 | HD | Denmark | 6 |
Vertebral fractures, which also impair quality of life, were seen on radiographs in 22%–28% of relatively young patients awaiting kidney and simultaneous pancreas–kidney transplantation . Not surprisingly, fractures occur more commonly in elderly patients, in women, in patients with diabetes, in those using glucocorticoids, and in patients with longer exposure to dialysis.
Earlier grades of CKD are also associated with an increased fracture risk . In CKD G3 and 4, hip fractures have been reported to be 2–3 times more common than amongst people without CKD . Age-standardized hip fractures were reported in 1.81/1000 persons with nondialysis dependent CKD versus 1.18/1000 for those with near normal kidney function, and in-hospital mortality, length, and costs of stay are all higher in the CKD patients . In Scotland, a review of medical records included all those with CKD G3–5 ( N =19,537) and a random sample of the population. After 5 years, the relative risk of hip fracture, after full adjustment for other factors, was significantly increased with an incidence rate ratio of 1.49 . In older men with CKD G3–5 receiving care from the Veterans Affairs, 15.7% experienced a fracture over 10 years. They also had elevated risk of death compared to those without CKD. Adjusting for competing risk showed that there remained a significant association between CKD and fracture . The Longitudinal Aging Study Amsterdam followed 1477 participants for 6 years, and those with CKD G3a and 3b had a 28% and 46% increase in the fracture risk, respectively, as compared to CKD G1 and 2 . The association of CKD to hip fractures is attenuated when competing risks of CKD-related mortality are taken into account .
Not all studies have found increased fracture risk with CKD G3–4 . One reason may be that creatinine does not accurately reflect renal function in patients who also have muscle disease or disuse. Therefore eGFR determined by cystatin C is more reliable, because cystatin C levels rise earlier in the course of CKD and do not depend on muscle mass . In elderly women and men those with higher cystatin C levels had significantly more fractures. In a more recent prospective cohort study of middle aged to elderly men and women followed for a median of 13 years, there was a stronger association of fracture risk with cystatin C than with creatinine, particularly in those with better renal function . A study of 988 Japanese women with osteoporosis (with exclusions including BMI<18 kg/m 2 , steroids, and rheumatoid arthritis, leaving 555 women for the analysis) found that those with lower GFR by cystatin C had higher fracture risk, but this was not seen when eGRF was measured using creatinine .
54.3.2
Coexistence of osteoporosis and chronic kidney disease
Early grades of CKD are common in elderly persons who have low bone mineral density, because both osteoporosis (defined as a T -score lower than −2.5) and CKD increase with aging. In the Third National Health Assessment and Nutritional Examination Survey, 84% of women with osteoporosis had CKD G3–4. The survey also found that 26% of women and 17% of men with CKD G3–5 had coexisting osteoporosis . These percentages varied with the age distribution of the population sampled.
54.4
Pathophysiology
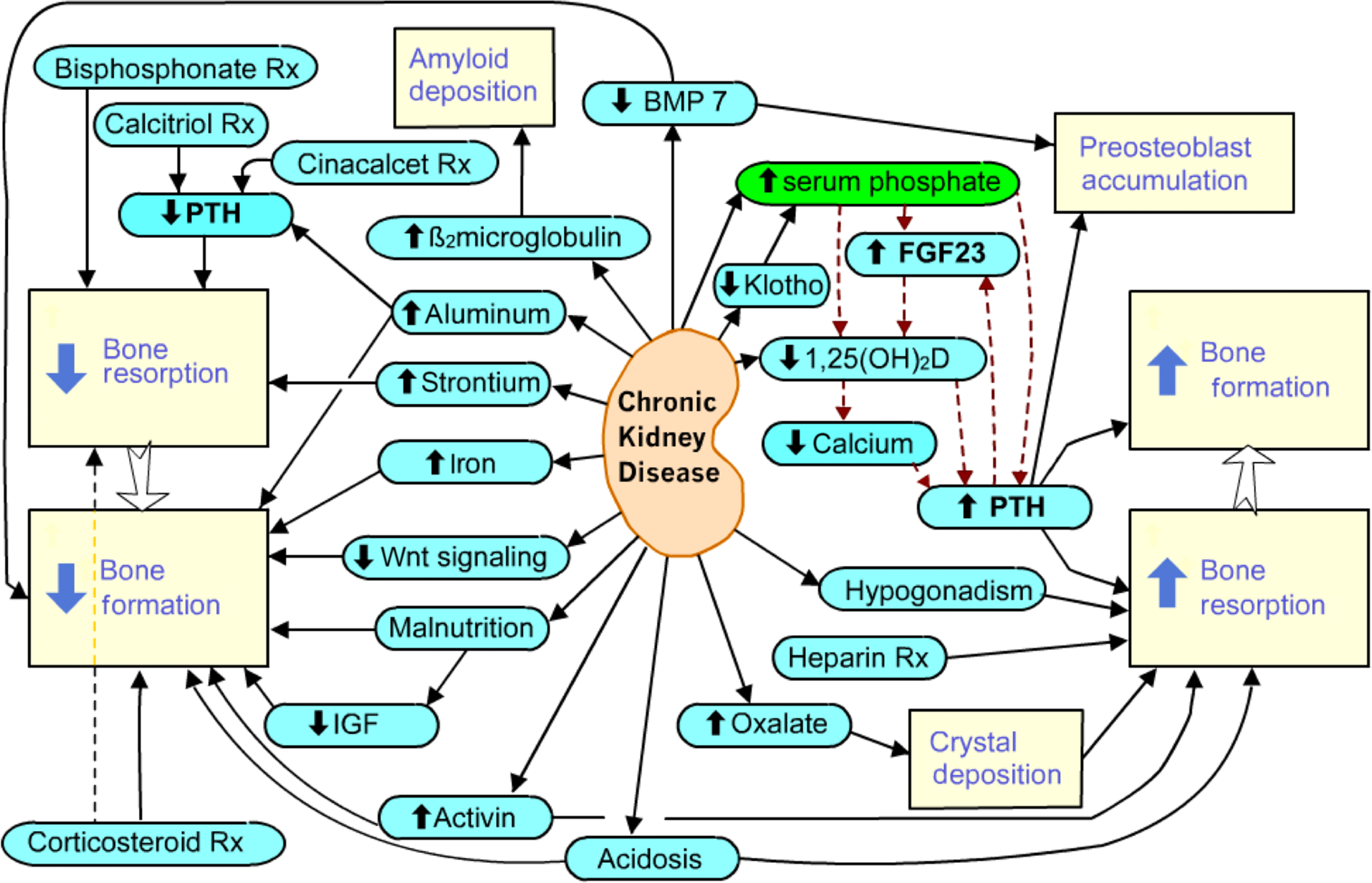
The pathophysiology of bone disease and mineral metabolism in renal failure is complex ( Fig. 54.1 ). The kidney plays an essential role in calcium, phosphate, and magnesium metabolism, not only by renal reabsorption and excretion but also by secretion of systemic hormones and growth factors. Changes in the serum mineral concentrations signal other systemic hormones that will act on bone cells. These changes result in a spectrum of bone disease that can prove difficult to predict for an individual patient.
54.4.1
Abnormal mineral metabolism
54.4.1.1
Overview
Calcium and phosphate balance is central to many metabolic pathways and the skeleton, kidney, and intestines work cooperatively to maintain mineral balance. To maintain homeostasis as GFR declines, the amount of a substance generated (or absorbed from the intestine) must equal the amount excreted. With progressive nephron loss, each intact nephron excretes a greater fraction of its filtered load. For example, while each nephron may need to excrete only 10% of its filtered phosphate load to maintain homeostasis at normal GFR, 90% may need to be excreted by residual nephrons at low GFR levels. Despite these adaptations, as CKD progresses the ability of remaining nephrons to excrete the daily load of phosphate is eventually exceeded, resulting in positive balance.
54.4.1.2
Phosphate
The phosphate bond is essential to life because it allows cells to control energy. Phosphorylation is necessary for glycolysis, enzyme activation, and DNA replication. Intracellular and extracellular concentrations of phosphate and calcium are tightly controlled by interrelated regulatory mechanisms, in which the kidney plays a pivotal role ( Fig. 54.2 ).
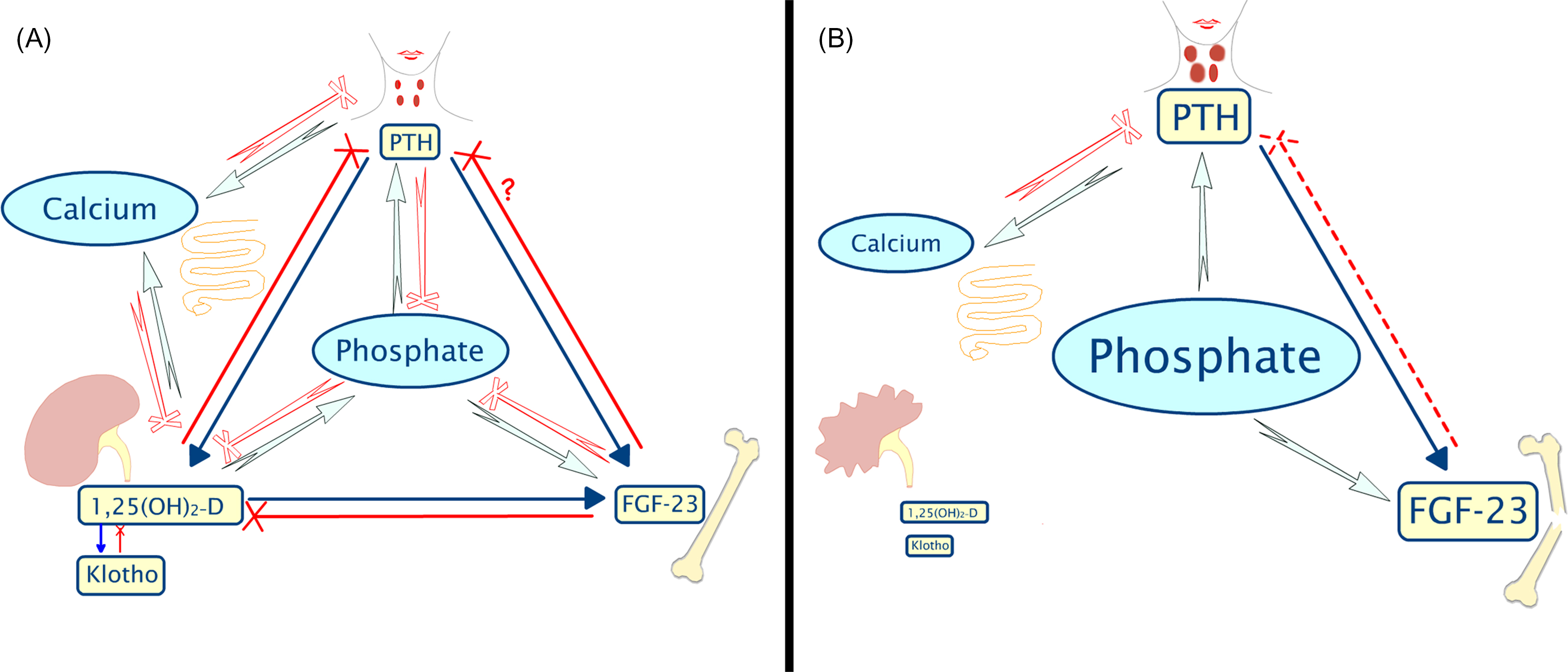
Active gastrointestinal absorption of phosphate is mediated by 1,25-dihydroxyvitamin D (1,25D). Phosphate excretion is predominantly renal and regulated by parathyroid hormone (PTH) and fibroblastic growth factor 23 (FGF23), both of which independently cause internalization and degradation of the proximal tubular sodium-dependent phosphate cotransporter (NPT)2a. Klotho, another hormone secreted in the nephron, is a coreceptor for FGF 23 and is necessary for its homing to target organs .
With progressive kidney failure, both FGF23 and PTH values rise in order to maintain phosphate homeostasis by increasing phosphate excretion from each remaining glomerulus. Consequently, elevated serum phosphate values are generally only seen in later stages of CKD G 4 and 5. Increased serum phosphate concentrations are directly associated with cardiovascular risk, mortality , and metastatic calcifications. Low phosphate levels are much less common in CKD, although hypophosphatemia may occur when patients undertake long dialysis hours, such as with nocturnal hemodialysis, or with malnutrition.
54.4.1.3
Calcium
Like phosphate, serum calcium concentrations are normally maintained within strict limits. High extracellular calcium can change membrane potentials necessary for cardiac, muscular, and neurological function. Low extracellular calcium predisposes to arrhythmias and muscle dysfunction, and chronically low calcium levels lead to osteoporosis or osteomalacia. Just as for phosphate, 1,25D increases the intestinal absorption of calcium, but unlike phosphate, calcium does not shift into cells, and any excess must be excreted by the kidneys or deposited in the skeleton or soft tissues. Calcium is not as plentiful in the diet as phosphate, and in the presence of CKD, low 1,25D levels result in low intestinal absorption, so typically the serum calcium levels are low. However, hypocalcemia stimulates PTH secretion and bone resorption with restoration of calcium levels, while over time, parathyroid hyperplasia can result in hypercalcemia.
Phosphate, calcium, and PTH are interrelated. An international study of the 5-year mortality in 35,721 patients analyzed combinations of factors and found the highest mortality in those with low PTH, target calcium, and high phosphate. The lowest mortality was in those with target PTH, low calcium, and target phosphate .
54.4.1.4
Magnesium
To maintain normal serum magnesium levels, around 100 mg (4.1 mmol) must be excreted daily by the kidneys. The principal storage sites for magnesium are muscle (20%) and bone (67%), where it is complexed with apatite crystals at a calcium to magnesium ratio of 50:1. About 20%–30% of total bone magnesium exists as a surface-limited ion, which is freely exchangeable with serum magnesium . Like other intracellular ions, serum magnesium levels do not necessarily reflect total body balance.
Most disturbances of magnesium metabolism result from renal or gastrointestinal magnesium losses, so CKD is unusual for being a cause of positive magnesium balance. However, hypomagnesemia can occur in CKD, related to drugs such as loop diuretics, calcineurin inhibitors, aminoglycosides, cisplatin, amphotericin B, and proton pump inhibitors and following parathyroidectomy or transplantation. Hypomagnesemia is also reported to be a significant predictor of cardiovascular and noncardiovascular mortality in patients undergoing hemodialysis .
Elevation of magnesium levels is generally caused by the use of Mg-containing antacids and phosphate binders and by the dialysate magnesium content. High Mg levels may inhibit mineralization of bone and reduced dialysate magnesium concentrations have been associated with an improvement in osteomalacia . However, long-term consequences of positive magnesium balance and the abnormal magnesium-to-calcium ratio found in bone biopsy studies remain unclear. Magnesium compounds are effective phosphate binders , which may reduce the development of arrhythmias and in observational studies of patients on dialysis, higher serum levels may have improved survival and slowed the development of coronary artery calcification (CAC) in patients with CKD G 3 to 4 .
54.4.2
Mineral-regulating hormones: parathyroid hormone, vitamin D, FGF23, and Klotho
The major circulating hormones that regulate mineral metabolism are involved in complex feedback networks, as shown in Fig. 54.2 . The direct consequences of renal disease include an inability to excrete phosphate or calcium. The kidney is the source of circulating Klotho and 1,25D, so those levels are decreased. This leads to increased concentrations of PTH and skeletal secretion of FGF23.
54.4.2.1
Parathyroid hormone
Secondary hyperparathyroidism is due to increased secretion of PTH as well as hyperplasia of the parathyroid glands. Initially, the hyperplasia is polyclonal but this changes to monoclonal and the cells may become resistant to suppression by calcium due to reduced calcium-sensing receptor (CaSR) expression. Finally, PTH levels may become very high and refractory to all nonsurgical treatments .
The progressive decline in levels of circulating 1,25D is a major contributor to hyperparathyroidism, because this hormone normally suppresses PTH levels by several mechanisms. 1,25D directly inhibits transcription of the PTH gene and exerts an antiproliferative effect on parathyroid cells by inducing expression of the cyclin kinase inhibitor p21 and decreasing expression of c -myc , which modulates cell-cycle progression from the G1 to S phase. 1,25D also upregulates expression of the CaSR , which sensitizes parathyroid cells to the inhibiting effects of serum calcium. 1,25D also indirectly reduces PTH secretion by increasing gastrointestinal calcium absorption. Finally, binding of 1,25D to the VDR and migration of this complex to the nucleus is reduced in the uremic state .
Low calcium levels also cause secondary hyperparathyroidism by stimulating PTH secretion via the CaSR. With persistent hypocalcemia the expression of the PTH gene is enhanced, due to greater stability of mRNA for PTH. When hypocalcemia is even more prolonged parathyroid hyperplasia ensues . Increased levels of serum phosphate also result in hyperparathyroidism by stabilizing PTH messenger RNA, and when prolonged, hyperphosphatemia reduces p21 expression and contributes to parathyroid gland hypertrophy .
A number of studies have addressed mechanisms by which the PTH mRNA transcript is stabilized. One mechanism involves the regulated binding of stabilizing AU rich binding factor 1 and destabilizing KSRP (K homology splicing regulatory protein), which is orchestrated by Pin 1 (peptidyl-prolyl cis – trans isomerase) . Another is by dysregulation of microRNAs, which regulate gene expression by controlling the binding of trans-acting proteins to mRNAs in order to influence their translation or degradation. Secondary hyperparathyroidism, whether induced by hypocalcemia or by uremia, has been associated with the dysregulation of a number of these miRNAs, leading to increased posttranscriptional PTH mRNA stability, and supporting a key role for miRNAs in the development of secondary hyperparathyroidism .
Reduced levels of 1,25D or high levels of dietary phosphate induce translocation to the cell surface of a tumor necrosis factor-α-converting enzyme (TACE), which results in release of the growth promoter TGF-α . In turn, TGF-α binds to and activates the epidermal growth factor receptor, which translocates to the nucleus where it functions as an autocrine signal for cell growth. With phosphate restriction, parathyroid TGF-α returns to normal levels rapidly, suggesting that lowering dietary phosphate may counteract uremia-induced parathyroid hyperplasia by preventing parathyroid TGF-α enhancement, in addition to inducing expression of p21.
Effect of fibroblastic growth factor 23 on parathyroid hormone
The effects of FGF23 on the parathyroid are unclear and may depend on duration of exposure or experimental conditions. Some experimental studies suggest that FGF23 inhibits PTH. But patients with CKD as well as patients with X-linked hypophosphatemic rickets consistently have both high FGF23 and high PTH.
Recently, Kawakami et al. found that mice with conditional knock-out (k/o) FGF receptors in the parathyroid glands had low PTH levels. This was true in mice with or without CKD. In cultured parathyroid cells without FGF receptors, there was no increase in PTH after 4 days of culture. When they added FGF23 to cultures of normal parathyroid cells, they found initial inhibition of PTH secretion, but after 4 days there was increased proliferation of the cells. They concluded that the long-term effect of FGF23 was to enhance circulating levels of PTH by causing parathyroid cells to proliferate.
Some experimental studies, however, have found that FGF23 inhibits PTH secretion . Administration of FGF23 to rats decreased PTH gene expression, and this was also seen in parathyroid cultures together with a slight increase in parathyroid cell proliferation . Another study in rats found that FGF23 inhibited PTH, and that blocking the FGF receptor completely restored PTH levels. But in acute hypocalcemia FGF23 did not inhibit PTH .
In patients with CKD the parathyroid glands appear resistant to any suppression by FGF23. This could be due to lower levels of Klotho , to time-dependent effects of FGF23, or to the fact that PTH secretion is controlled by several pathways.
Conversely, PTH increases secretion of FGF23. This is a consistent finding (discussed in section 54.4.2.3 ).
54.4.2.2
Vitamin D
A decline in levels of 25-hydroxyvitamin D (25(OH)D) often accompanies progressive CKD . Low levels of 25(OH)D are inversely related to PTH concentrations from CKD G1 to 4 . This is not necessarily due to the kidney disease itself. On the other hand, 1,25D levels consistently decline even in early CKD when proximal tubular function should still be adequate to hydroxylate the 25(OH)D. This is due to an early rise in FGF23 levels that inhibit 1-alpha-hydroxylase (CYP27B1) activity and induce 24-hydroxylase activity (CYP24A1), resulting in decreased production and increased breakdown of 1,25D. This may be exacerbated by low 25(OH)D levels. With worsening CKD, proximal tubular damage reduces the ability of tubular cells to produce 1,25D. Low 1,25D levels are responsible for hypocalcemia, hyperparathyroidism, and many of the skeletal manifestations of CKD patients.
54.4.2.3
Fibroblastic growth factor 23
FGF23 is an endocrine hormone with multiple actions ( Table 54.4 ), many of which depend on signaling through the FGF1 receptor which must be associated with Klotho. The major effect is to decrease serum phosphate levels.
| FGF23 | FGF23+membrane Klotho | Soluble Klotho | ||
|---|---|---|---|---|
| ⇧ Cardiac hypertrophy | ⇧ Renal PO 4 excretion | ⇩ Intestinal PO 4 absorption | ||
| ⇩ Immunity | ⇩ Cyp27B | ⇧ Longevity | ||
| ⇧ Liver inflammation | ⇩ Wnt signaling (Dkk1 secretion) | ⇩ Wnt signaling (Binds to wnt) | ||
| ⇩ Sodium excretion | ⇩ Calcium excretion | |||
| ? Effects on Klotho * | ⇧ FGF23 | |||
| ? Effects on PTH secretion | ⇩ PTH | |||
| ⇩ ACE | ⇩ Vascular calcification (Blocks cell differentiation) | |||
| ⇩ TGF-β signaling | ||||
* FGF23 decreases Klotho . FGF23 elevates mKlotho and cKlotho . FGF23 increases or decreases Klotho . Klotho not regulated by FGF23 . ACE, α-Converting enzyme ; PTH, parathyroid hormone .
Fibroblastic growth factor 23 metabolism
FGF23 is a protein with 251 amino acids expressed mainly in the osteocytes . The N-terminal end binds to the FGF1 receptor and the C-terminal end interacts with Klotho. Between are several O -glycosylation sites where FGF23 is cleaved within the Golgi apparatus. The FGF23 is cleaved by the protein furin and other protein convertases. This cleavage is important to maintain physiologic levels of FGF23 . In addition, the C-terminal FGF23 can act as a competitive antagonist to FGF23 .
GALNT3 ( N -acetylgalactosaminyltransferase 3) stabilizes the FGF23 at these sites to maintain higher levels of intact FGF23. High phosphate increases GALNT3 activity and therefore increases intact FGF23. Conversely, FAM20C (family with sequence similarity 20 member C) phosphorylates FGF23 and prevents GALNT3 from stabilizing the FGF23. In patients with CKD, there is dysregulation of the cleavage, so more FGF23 is in the active intact state .
Multiple different factors modify FGF23 levels ( Fig. 54.3 ). Although the main driving factor for FGF23 production is a chronic increase in phosphate load, the phosphate itself does not directly increase FGF23 secretion. However, low phosphate is detected by the PIT-2 transporter, which inhibits FGF23, and high phosphate increases GALNT3 activity .
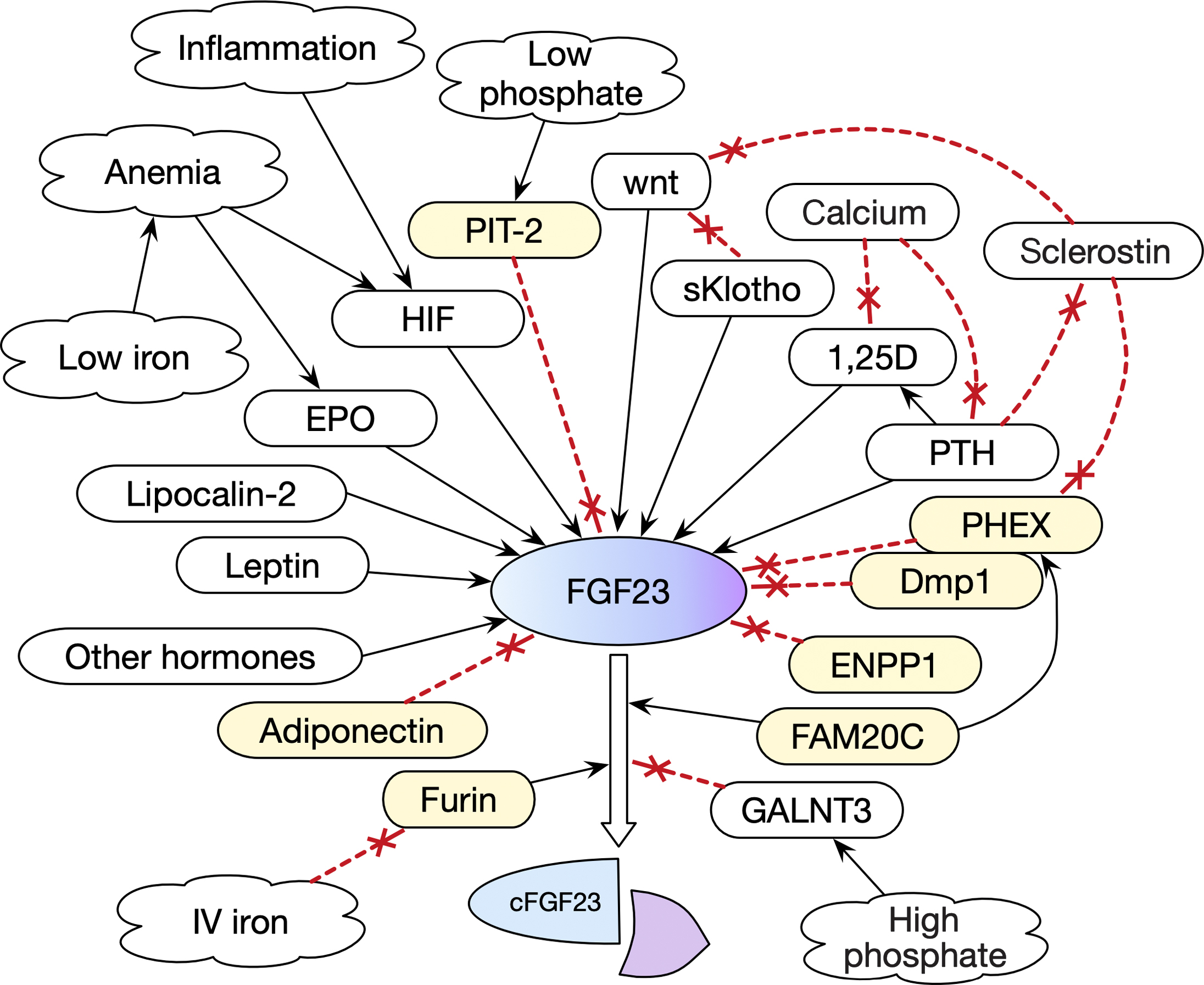
Proteins associated with the cell surface, including PHEX and Dmp-1, regulate mineralization and inhibit FGF23 synthesis . ENPP (ectonucleotide pyrophosphatase phosphodiesterase) synthesizes pyrophosphate, a potent extracellular mineralization inhibitor, and also inhibits FGF23.
Circulating hormones increase FGF23. 1,25D acts through osteocyte vitamin D receptors to increase FGF23 transcription . Treatment of patients with vitamin D analogs had a net effect of increasing the FGF23 levels . PTH increases FGF23 both directly and indirectly. PTH directly activates the PKA pathway in osteoblasts, which increases FGF23 mRNA levels. In mice with constitutive activation of PTH receptor signaling in osteocytes, there is elevated FGF23 expression . PTH indirectly increases FGF23 by converting 25(OH)D to 1,25D, which increases FGF23 . On a clinical level, lowering PTH levels in hemodialysis patients with cinacalcet or parathyroidectomy will decrease FGF23 levels.
Wnt-signaling increases the expression of FGF23. Umr-106 osteoblast-like cells stimulated with β-glycerophosphate (β-GP) increase expression of FGF23 as well as markers of intracellular wnt-signaling. The FGF23 increases further with addition of lithium, which enhances wnt-signaling . In UMR 106 osteoblast-like cell culture, adding sclerostin (which inhibits wnt-signaling) abolished the ability of PTH to increase FGF23 . Mice with PTH receptor activation express excess FGF23, but this is inhibited by sclerostin . Sclerostin has dual, opposing effects because it also inhibits PHEX which results in greater secretion of FGF23 .
Soluble Klotho also has dual opposing effects of decreasing FGF23 via inhibiting wnt and increasing FGF23 via the FGF receptor signaling pathway. In UMR 106 cells that were cultured with β-GP, addition of Klotho attenuated the increase in both FGF23 and the markers of wnt signaling. This was reversed with lithium . However, mice that were infected with an adenovirus producing soluble Klotho developed high FGF23 along with hypophosphatemia, severe osteomalacia, hyperparathyroidism, hypocalcemia, and low 1,25D. The investigators thought the increase in FGF23 was potentially through the Klotho–FGFR–FGF23 receptor complex . The same constellation of findings was seen in a patient with a mutation that increased Klotho secretion .
Several other factors play a role in FGF metabolism . Leptin induces FGF23 in rat calvarial cultures and in ob/ob mice . Adiponectin inhibits FGF23. Estrogens also have been shown to dose-dependently increase FGF23 levels through transcriptional upregulation . Aldosterone also increases FGF23. TNF alpha and IL-6 increase FGF23 transcription in oocytes . When an enhancer of FGF23 was deleted from thymus and spleen FGF23 values were lower .
A recently discovered factor that directly increases FGF23 is lipocalin-2, which is a proinflammatory and iron-shuttling molecule expressed by the kidneys in response to acute inflammation .
Fibroblastic growth factor 23 and iron deficiency
Erythropoietin, iron deficiency, and hypoxia-inducible factor also stimulate FGF23 . Iron deficiency increases FGF23 secretion even in normal persons , but the protein is degraded so that the intact molecule levels return toward normal, the fragments are increased, and the serum phosphate remains normal. In CKD G1–4 the serum iron was inversely correlated to serum FGF23 using an assay that measured both intact and C-terminal fragments . In acute kidney injury, erythropoietin increases secretion of FGF23 from bone marrow cells via an erythropoietin receptor .
Paradoxically, iron infusions increase FGF23 in dialysis patients and some degree of hypophosphatemia occurs in 2.1%–32.1% of patients without CKD after intravenous ferric polymaltose or carboxymaltose for treatment of iron deficiency . It is possible that iron infusions inhibit furin, and consequently, stabilize intact FGF23 levels .
Fibroblastic growth factor 23 actions
FGF23-Klotho signaling decreases renal phosphate reabsorption in the proximal tubule by causing internalization and proteolytic degradation of the sodium-phosphate transporters ( Table 54.4 ). FGF23-Klotho also inhibits Cyp27B1 (1-alpha-hydroxylase) as well as increasing the activity of the catabolic 24-hydroxylase enzyme (CYP 24A1). This latter action enhances the conversion of 1,25D to 1,24,25(OH) 3 D and reduces the availability of substrate to the 1-alpha-hydroxylase enzyme by converting 25(OH)D to 24,25(OH) 2 D . The low 1,25D levels will decrease the serum phosphate because there is less absorption from the intestine.
Reports about the effects of FGF23 on production of Klotho are inconsistent . Some find increased Klotho and others do not . There may be indirect effects because FGF23 decreases 1,25D which stimulates expression of Klotho.
In the distal nephron , FGF23-Klotho increases calcium and sodium reabsorption by activating ANK4, which increases the epithelial calcium channel transient receptor potential vanilloid-subtype 5 (TRPV5) and decreases angiotensin-converting enzyme. These actions contribute to the left ventricular failure seen in patients with CKD .
In the heart , FGF23 reduces contractility and enhances arrhythmogenicity in cardiomyocytes by mishandling intracellular calcium . Cardiac FGF23 signaling through the FGFR4 activates the calcineurin-NFAT signaling pathway to increase left ventricular hypertrophy (LVH) . Pharmacological interference with cardiac FGF23/FGFR4 signaling can protect or even reverse worsening LVH in a rat model of CKD . Patients with X-linked hypophosphatemia also have high FGF23 levels but do not develop LVH, and recent studies suggest that the hypophosphatemia protects the heart against the actions of FGF23 .
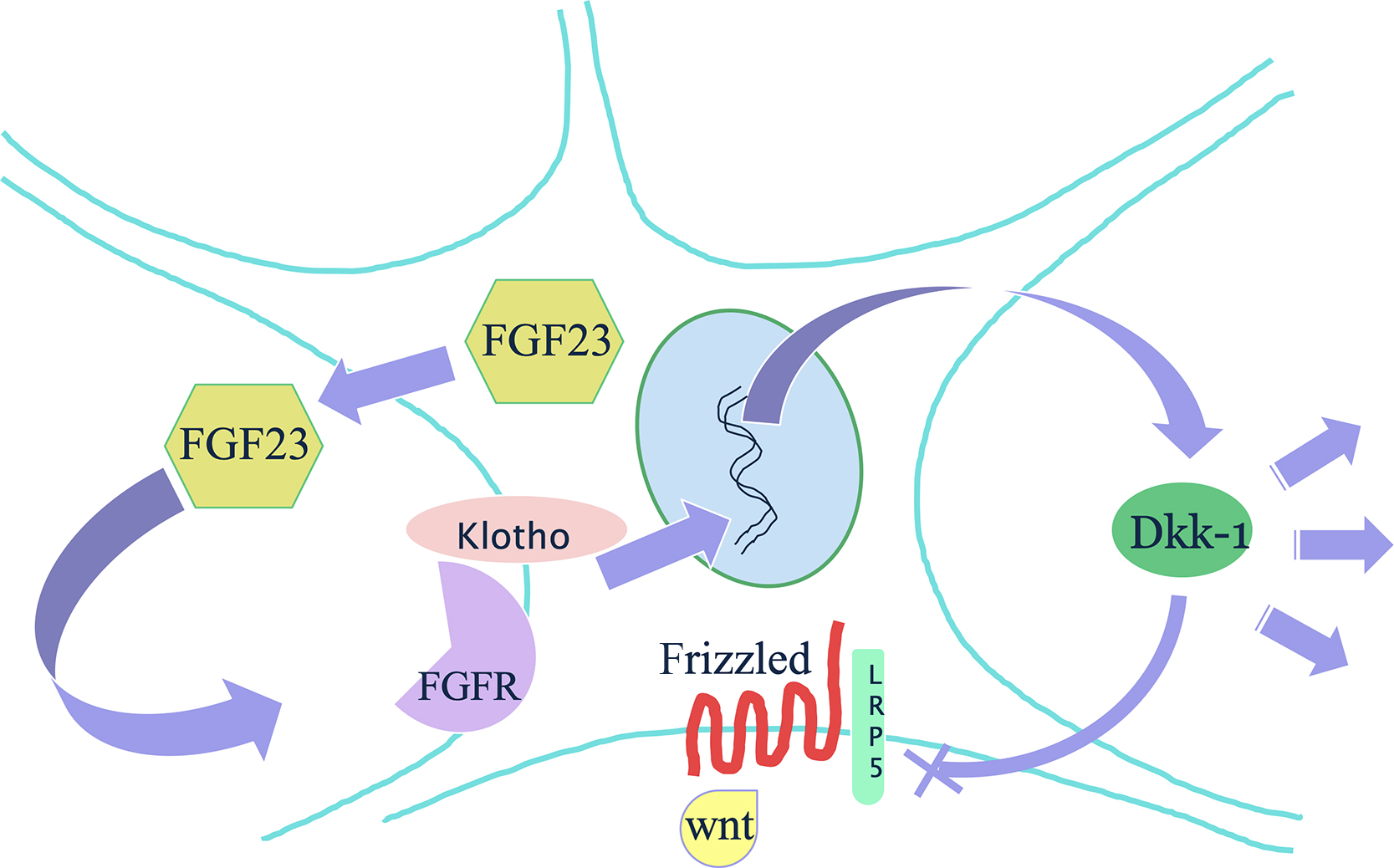
In the bone , FGF23 has an autocrine effect of inhibiting the wnt-signaling pathway ( Fig. 54.4 ). In uremic rats fed a high phosphate diet, FGF23 and sKlotho serum levels increased, and in the bone, there was an increased expression of Dkk1 and Sfrp1. The rats subsequently lost bone. Further investigations in an osteoblast cell line exposed to uremic serum found inhibited mineralization and proliferation . When osteoblast-like cells were exposed separately to serum with high PTH, FGF23 or sKlotho, there was no increase in Dkk1, but the combination of FGF23 and sKlotho was what increased Dkk1 and thereby inhibited wnt-signaling A direct action of FGF23 on bone mineralization is independent of effect on systemic phosphate . Also, FGF23 is a potent suppressor of alkaline phosphatase (ALP) .
Fibroblastic growth factor 23 knock-out mice
FGF23-k/o mice have hyperphosphatemia and multiple abnormalities, including emphysema, thickening of vessel walls with extensive calcifications, amyloid accumulation in heart and kidneys, hypogonadism, atrophic skin, reduction in bone density, and premature lethality . They also have high 1,25 vitamin D levels and high osteopontin levels, with osteomalacia . Mice with double k/o of FGF23 and 1a(OH)ase did not have the atherosclerosis, soft-tissue calcifications or short life span of the FGF23 k/o mice , but the bone density in the double k/o mice was even lower than in the FGF23 k/o mice ( Table 54.5 ).

Fibroblastic growth factor 23 in chronic kidney disease
An increase in FGF23 levels is detected in very early CKD. In otherwise healthy kidney donors, FGF is increased despite decreased serum phosphate levels . Similar results were seen in another study of 3879 patients with CKD G2–4. The mean serum phosphate and median PTH levels were still in the normal range, but median FGF23 was markedly greater than in healthy populations, and increased significantly with decreasing eGFR . In children with CKD, levels of FGF23 are also reported to increase prior to the development of hyperphosphatemia . In acute kidney injury after cardiac surgery, FGF23 levels rose 15.9-fold in 1 day .
In a mouse model of renal injury the serum FGF23 levels were increased prior to elevations in BUN and serum creatinine, but the gene transcription in osteoblasts was not increased until CKD further declined. The source of the early increase in FGF23 was not clear, but the authors thought it could represent changes in excretion or posttranscriptional processing of the hormone . A different acute kidney injury model also found rapid increase in FGF23 along with increased bone production . Most of the FGF23 in patients with CKD is intact , suggesting that there may be something in uremic serum that inhibits the cleavage of FGF23 .
Patients with polycystic kidney disease have higher FGF23 levels than those with other kinds of CKD. Most of the FGF23 is cleaved, so they do not have excess phosphaturia. The source of this FGF23 is the liver, and surgical reduction of the cystic liver mass is associated with a decrease in the FGF23 levels. This is independent of GFR, phosphate, or iron .
Fibroblastic growth factor 23 levels and clinical bone disease
In patients on hemodialysis, FGF23 values did not correlate to bone mass, or to serum levels of bone-specific ALP or C-telopeptide . However, in CKD G 1–4, FGF23 levels were inversely correlated with bone mineral density at the hip . A study of 105 Japanese patients with CKD found that those with vertebral fractures had higher FGF23 levels . In a case-control study from a cohort of 268 Canadian dialysis patients, those with fractures had higher FGF23 . The Health ABC study followed 2234 patients with serial bone density measurements and fracture outcomes. In those without CKD the FGF23 levels were not related to bone density changes, but in people with CKD, higher FGF23 levels were associated with greater bone loss. However, fracture risk was not associated with FGF23 .
Fibroblastic growth factor 23 levels and other clinical outcomes
High FGF23 concentrations are associated with poor clinical outcomes, including cardiovascular disease, both in patients with renal disease and in the general population . Compared with the lowest quartile of FGF23, each subsequent quartile was associated with a progressively higher risk for death. In the MESA study of older adults, FGF23 was associated with heart failure with normal ejection fraction . However, it is not clear if these problems are caused by FGF23 or if it is a surrogate marker for disease. A metaanalysis of studies relating FGF23 to cardiovascular disease did not find dose relationships and the authors concluded that the association was not causative .
In patients with CKD and hyperparathyroidism, a 30% or more reduction in FGF23 induced by cinacalcet was associated with lower rates of cardiovascular mortality than seen with placebo . Within this study, a polymorphism in FGF23 was associated with cardiovascular mortality .
54.4.2.4
Klotho
Discovery
In 1997 when Kuro-o was searching for genes that altered the life span of mice, he discovered a gene that increased life span when overexpressed, but with an inactivating mutation, the mice had shortened life span and many features that are also seen with aging (arteriosclerosis, emphysema, thin skin, infertility, kyphosis). He named the gene Klotho after the first of the Moirae, who were the three Greek goddesses controlling man’s fate. Klotho spun the thread of life .
Metabolism
Klotho is a 130-kDa single-pass transmembrane protein expressed in the nephron, the parathyroids, and the choroid plexus. Although expression is highest in the distal nephron, Klotho is also found in the proximal tubule and in the bone at very low levels .
The Klotho promoter contains response elements for vitamin D and for thiazolidinediones. 1,25D increases secretion of Klotho , whereas aldosterone, angiotensin II, and adiponectin suppress transcription . Inflammation or injury can increase micro-RNA34 in the renal tubules, and this results in decreased Klotho . It is not clear whether FGF23 directly decreases Klotho synthesis, or whether there is indirect inhibition due to lower 1,25D. In rats with unilateral obstruction, Klotho expression decreased in the diseased kidney but did not change in the normal kidney, suggesting that the Klotho reduction was due to local factors .
The Klotho ectodomain can be cleaved by ADAM10 and released as a soluble form of Klotho. Insulin may stimulate this process .
Klotho actions
Klotho is both a coreceptor and a systemic hormone. Both membrane Klotho and soluble Klotho associate with FGF1 receptors to allow FGF23 signaling . Soluble Klotho acts as a membrane scaffold to tether FGF23 to the FGF1 receptor , but it is not clear if that complex signals as well as the complex with membrane Klotho .
Klotho-FGF23 signaling in the proximal tubule will affect bone by reducing serum phosphate and 1,25D. Soluble Klotho also has effects that are independent of FGF23. In the distal nephron, Klotho can increase calcium reabsorption. Previously, it was thought that this action was related to glucosidase activity (Klotho would trap the TRPV calcium channels in an open position) but new studies of the molecular structure of Klotho show this is not correct. Instead, soluble Klotho binds to ganglioside-enriched lipid rafts, which allows greater calcium reabsorption .
Soluble Klotho can inhibit important signaling systems at the membrane level, including wnt, TGF-β, and PTH. In vascular smooth muscle cells (VSMCs) Klotho inhibits wnt signaling, thereby preventing arterial calcifications. This effect is reversed with lithium . In cultured bone cells (osteoblast-like UMR-106), soluble Klotho binds to wnt, thus inhibiting the wnt-signaling pathway and decreasing FGF23 secretion .
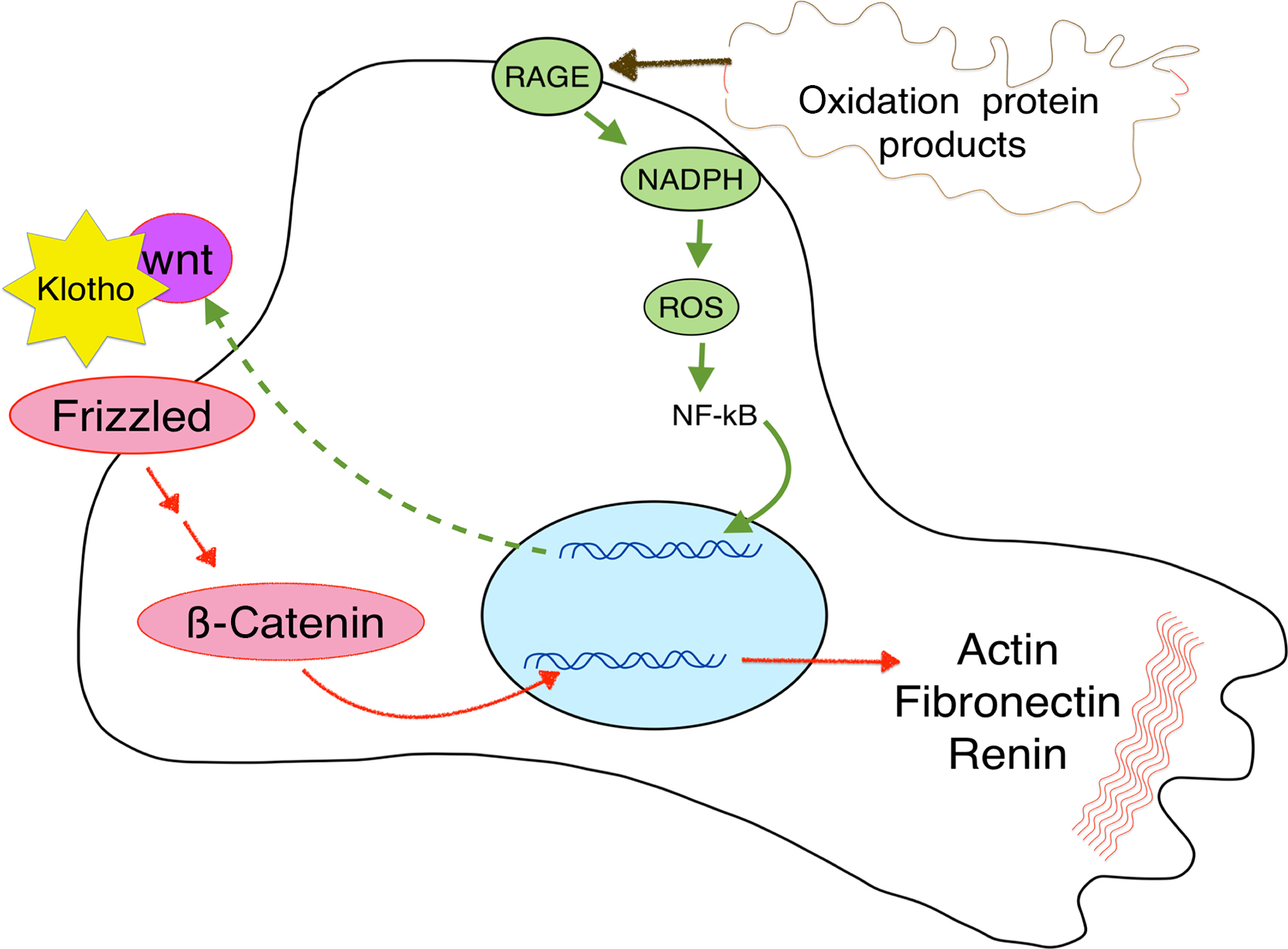
Klotho can also block wnt signaling in the kidney ( Fig. 54.5 ), thereby preventing renal fibrosis .
Knock-out mice
Table 54.5 shows phenotypic features of genetically altered mice. 1,25D levels are elevated in Klotho −/− mice . Double k/o mice (Klotho and VDR) had hypophosphatemia and low serum FGF23, with no soft-tissue calcifications, emphysema, or skin changes .
The Klotho mutated ( kl/kl ) or k/o mice resemble the FGF23 −/− mice in most systems but there are differences in the skeleton. Although the kl/kl mice have kyphosis and thin cortical bone, they have increased trabecular bone. This pattern is typically seen in patients with CKD who have hyperparathyroidism, but in the kl/kl mice the PTH was not increased. Histology showed low numbers of both osteoblasts and osteoclasts . High-resolution micro-computed tomography (microCT) found that the trabecular bone was denser than in wild-type mice . In addition, the bone was resistant to the bone loss that is seen with unloading . When the kl/kl mouse was crossed with mice heterozygous for deletion of osteoprotegerin (OPG), the metaphyseal region no longer showed dense trabecular bone . Further studies demonstrated that this high trabecular bone density was seen because Klotho inhibits wnt-signaling. Therefore the Klotho k/o mice had more β-catenin in the osteoblasts, which leads to increased bone formation, as well as increased OPG .
Conditional knockout of Klotho in the osteocytes results in increased bone formation and increased density . This is also consistent with the findings that Klotho inhibits wnt signaling.
The findings are more complicated when Klotho k/o mice develop uremia. The complex interactions of metabolism between FGF23 and Klotho were explored in a study in which mice were generated with conditional Klotho k/o in mesenchymal cells (which develop into bone of the limbs) who underwent 5/6 nephrectomy. When they developed uremia, they increased transcription of FGF23 from the axial skeleton but not from the limb bones .
Klotho in chronic kidney disease
In early renal failure Klotho is decreased even though there are still enough nephrons to maintain levels. This could be due to the reduced 1,25D caused by rising levels of FGF23. In a study of dialysis patients, those with osteoporosis at the femoral neck had lower serum klotho levels than those with osteopenia or normal bone density, whereas at the lumbar spine there was no difference between those with osteoporosis or osteopenia .
Klotho and cardiovascular disease
In animal models of CKD, enhanced expression of soluble Klotho results in fewer vascular calcifications . Klotho inhibited VSMC transformation and this was partially mediated by inhibition of wnt pathway .
Several studies have shown that soluble Klotho may be cardioprotective. In a 6-year clinical trial that enrolled 3555 subjects with heart disease, those with low Klotho at baseline had more heart failure or mortality. Those with low Klotho and high FGF23 had a 16.9% rate of CV death or heart failure compared to 3.4% in high Klotho-low FGF23 group . In a study of 2010 patients with CKD G 1–5 from Korea, the left ventricular mass showed an inverse association with serum Klotho. The study excluded patients with serious heart disease and adjusted for phosphorus and FGF23 .
54.4.3
Signaling pathways: Wnt, activin
54.4.3.1
Wnt-signaling pathway
In 2001 identification of the high bone mass gene as LRP-5, a coreceptor for the wnt-signaling pathway, led to numerous studies that documented the importance of this pathway in skeletal metabolism . Sclerostin and Dickkopf (Dkk1) are two soluble proteins that inhibit wnt signaling by binding to LRP-5. Secreted frizzled-related proteins (sFRP4) also inhibit the pathway by acting as decoy receptors. Dkk1 is expressed by many kinds of cells, including those in the bone, kidney, marrow, and vasculature.
Sclerostin is expressed almost exclusively in mature osteocytes. It binds to LRP 5/6 and LRP4 on immature osteoblasts which inhibits anabolic wnt signaling and prevents inchoate osteoblasts from maturing. Sclerostin may also reduce in vitro mineralization . When the osteocytes sense mechanical loading, they stop secreting sclerostin, thus allowing wnt signaling to progress . Mutations that inactivate the sclerostin gene cause dense bones due to enhanced bone formation, but people with heterozygous mutations are healthy and rarely have fractures. Romosozumab is an antisclerostin antibody that markedly increases bone formation in mice and humans, and has recently been approved by the Federal Drug Administration for treatment of osteoporosis. In a rat model of renal osteodystrophy, it has also been reported to increase bone density and strength .
Serum sclerostin and Dkk1 levels in chronic kidney disease
Serum sclerostin values increase with reduction in eGFR and in patients on hemodialysis, sclerostin values are approximately fivefold higher than in premenopausal women without CKD, and twofold higher than postmenopausal women . In a cross-sectional study of patients with CKD, urinary excretion of sclerostin was around 10-fold higher in patients with CKD G5 than in patients with CKD G1 , and in older adults from Iceland, those with an eGFR<45 mL/min/1.73 m 2 had higher sclerostin than those with eGFR>60 mL/min/1.73 m 2 . They also had more bone marrow fat . The increased sclerostin may have a protective effect. A 4-year study of 396 dialysis patients found higher sclerosin levels associated with lower mortality .
PTH decreases sclerostin expression, and as patients develop hyperparathyroidism the sclerostin decreases . In a study of 88 patients with CKD G3–4 who were treated with the vitamin D receptor activator paricalcitol, levels of sclerostin increased, which was likely due to the reduction in PTH .
With progression of CKD-MBD, osteocytes secrete more FGF23, which has an autocrine action to increase osteocytic secretion of Dkk1 . Serum levels of Dkk1, however, are not consistently elevated in clinical studies. In an observational study of 308 patients with CKD the Dkk1 was lower than in controls .
wnt signaling in bone in chronic kidney disease
In a mouse model, an early increase in expression of SOST led to reduced wnt signaling and a consequent increase in the RANKL/OPG ratio .
FGF23 has an autocrine effect to inhibit wnt-signaling in bone. In a study of rats with CKD a high phosphate diet resulted in high FGF23 and PTH. Sclerostin increased at 8 weeks but then decreased. Meanwhile Sfrp1 and Dkk1 increased with time. Further studies in UMR cells exposed to uremic serum, or to FGF23 and Klotho, showed increased expression of Dkk1 and Sfrp1 (see Fig. 54.4 ).
Patients in different grades of CKD show increasing expression of sclerostin by the osteocytes, which is associated with lower bone formation . Higher turnover was seen in patients with high PTH and low sclerostin .
In bone biopsies from 60 patients with CKD G5, Cejka et al. investigated the associations between serum levels of sclerostin or Dkk1 and histomorphometric parameters of bone turnover, mineralization, and volume (TMV). Serum sclerostin levels were higher in CKD patients than in normal women. Sclerostin correlated negatively with histomorphometric parameters of turnover, osteoblastic number, and function in both unadjusted and adjusted analyses. Furthermore, sclerostin was superior to PTH for positive prediction of high bone turnover. However, Dkk1 did not show correlations with any of the parameters of bone turnover measured on bone biopsies.
Sclerostin has been studied in relationship to bone density in CKD patients, with inconsistent results. In 89 dialysis patients, sclerostin correlated inversely with bone mineral density (BMD) and cortical mass changes .
wnt signaling in kidney
Although wnt signaling is beneficial to the skeleton, it plays a negative role in other systems. In the kidney, wnt signaling is active during development but becomes low in adults. In most experimental animal models of kidney injury or CKD, including kidney aging, wnt is upregulated . This response to injury seeks to recapitulate growth to repair damage, but it can result in fibrosis and worsening renal disease. For example, in the kidneys wnt signaling leads to podocyte dysfunction, proteinuria , and increased epithelial damage . When podocytes were exposed to oxidative stress, wnt-signaling was increased. If this signaling is blocked by either Klotho or knockdown of β-catenin, the podocyte damage was reduced ( Fig. 54.5 ). Wnt production in the renal cells has autocrine functions, but the signaling also increases Dkk1 secretion, which can enter the circulation and decrease the bone formation rate.
wnt signaling in vasculature
Wnt signaling also has vascular effects. In VSMCs, activation of wnt signaling promotes the transition to an osteoblast phenotype. The cells react by secreting Sfrp4 . In a rat model, of 5/6 nephrectomy, aortic sclerostin mRNA was increased .
Sclerostin-k/o (SOST −/− ) mice with normal kidneys had higher carotid artery calcification than wild-type, but these were not significant (although the mean was five times as high). When the SOST −/− mice and controls underwent 5/6 nephrectomy the survival time was similar and the degree of renal failure and increase in PTH and degree of vascular calcifications were also similar, but there was higher cardiac calcium content in the SOST −/− mice. In the bones the SOST −/− sham and nephrectomized mice had much greater bone area than controls. They also had higher osteoblast perimeter and bone formation rates .
In a clinical study of 67 patients on dialysis, CACs measured by CT showed significant correlations with serum sclerostin levels. Analysis of calcified aortic valves in 10 dialysis patients revealed strong sclerostin expression which was not seen in noncalcified valves .
Whether the circulating sclerostin has a vascular effect is uncertain, but this could be important, because treatment of bone disease with antisclerostin medications could potentially increase vascular calcifications . A study of 157 kidney transplant patients reported that baseline sclerostin levels were associated with cardiovascular calcification, but sclerostin levels did not predict cardiovascular events, fracture, or mortality over the next 3.7 years .
54.4.3.2
Activin
Activin is a member of the TGF-β family. It increases with renal injury and is involved in both skeletal disease and vascular calcification. This signaling pathway leads to transcription of various genes depending on the system and the conditions and is highly complex because of crosstalk between different ligands . Activin binds primarily to the type A receptor. The type II receptors then activate the type I receptor (activin receptor–like kinase, or ALK). This leads to phosphorylation of SMAD 2 and 3, which translocate to the nucleus and activate transcription . Chronic activation of the activin pathway favors transcription of ubiquitin ligases and impairs Akt/mTOR-mediated protein synthesis .
Activin and the skeleton
The effects in the skeletal system vary depending on the physiological conditions. Activin is embedded within the bone matrix and is released by resorption, as is TGF-β. Giving activin to ovariectomized rats increases bone formation and bone density, but only at low doses . Some studies have found stimulation of osteoblasts, others inhibition of bone formation . Likewise, both increases and decreases in osteoclasts have been reported . In osteoblast cultures, activin A inhibits mineralization . In a mouse model of disuse osteoporosis, activin receptor blockade mitigated the loss of femoral neck bone mass . Primates treated with an inhibitor of activin signaling show both increased bone formation and decreased bone resorption , and a small randomized clinical trial in women with osteoporosis showed increases in markers of bone formation and decreased markers of bone resorption .
Activin signaling in the kidney
In kidney, activin A levels increase, and trigger proliferation of fibroblasts, following ischemia or injury . Ureteral ligation increased activin A plasma levels as well as expression in the obstructed kidney but in the normal kidney there were no changes .
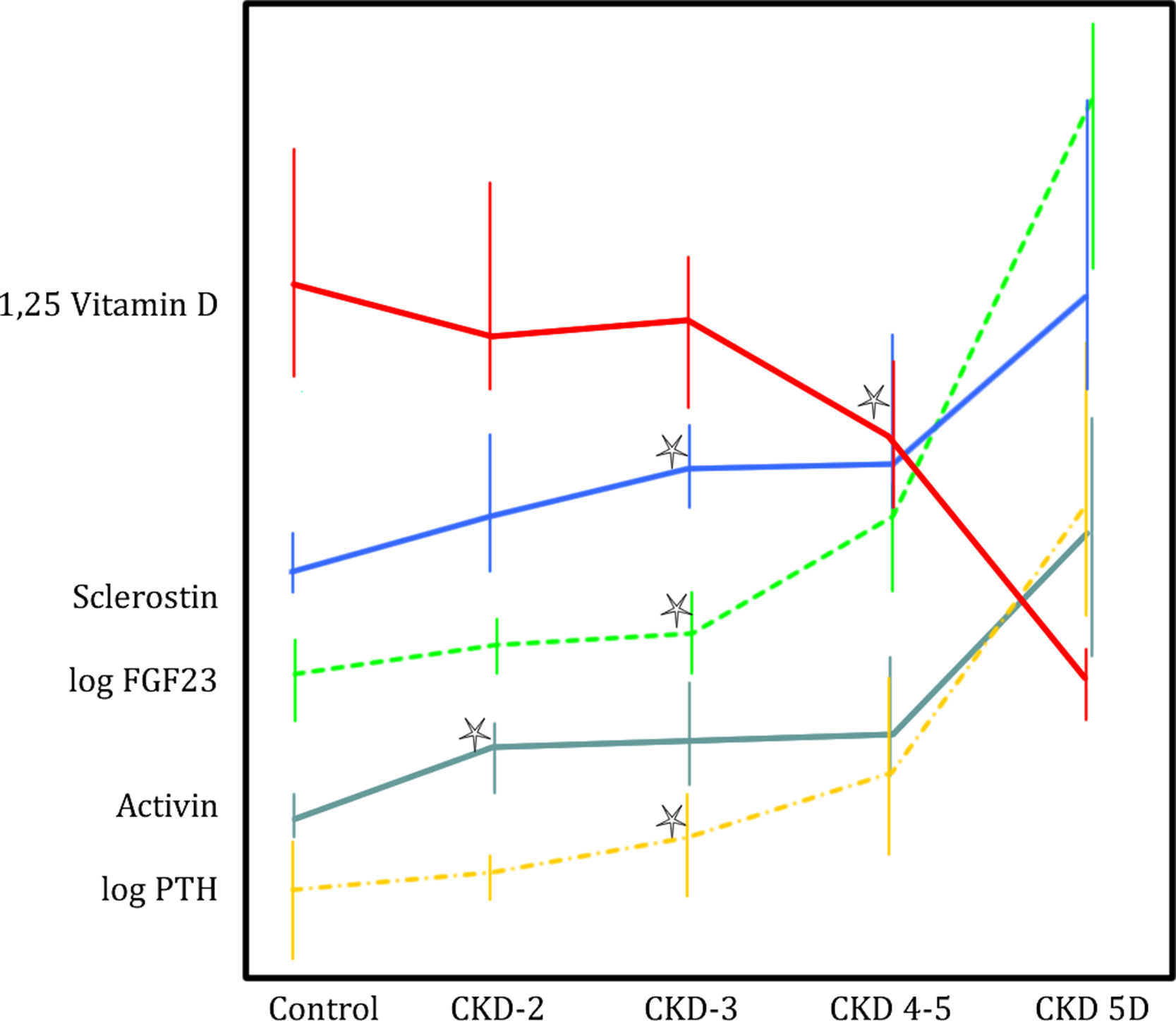
In 104 patients with CKD G 2–5, serum activin A increased significantly in CKD G 2, whereas PTH and sclerostin were not increased until CKD G 3, and FGF23 and 1,25D were not significantly changed until CKD G 4. The activin A levels were higher in those with high bone turnover and levels showed significant correlations to both the bone formation rate and the osteoclast surface ( Fig. 54.6 ).
Activin signaling in the vasculature
Activation of the ACRII receptors can increase VSMC transformation into osteoblast-like cells that can cause medial calcifications. This happens early in CKD . Activin-A can inhibit bone morphogenetic protein (BMP) 7 signaling in the vasculature, because there is competition for Smad4 . Since BMP-7 inhibits vascular calcification, the effect of activin-A is to promote the calcification. On the other hand, exogenous BMP-7 can inhibit the aortic expression of activin-A .
Effects of blocking activin signaling
Soluble activin RIIA is a ligand trap, and this mitigates bone loss in a mouse model of disuse osteopenia . In young mice, skeletal muscle hypertrophy was seen with administration of a ligand trap .
Treatment with an ActRIIA ligand trap in Alport mice reversed aortic expression of osteoblast transition markers Runx2 and osterix. The mice did not develop cardiac hypertrophy. Bone histomorphometry showed reductions in resorption as well as increases in bone formation rate. The treated mice and control mice had similar bone-forming surfaces and osteoblast numbers, but the mineral apposition rate was higher in treated mice, indicating greater forming activity per osteoblast. The inhibition of osteoclastogenesis was distal to RANK activation; thus it could block osteoclasts even when PTH was present . Renal function did not deteriorate as quickly as in the control mice. Similar results were seen when the ligand trap was used in other mouse models of CKD . In ldlr −/− mice with CKD, sKlotho levels were increased with the ligand trap .
The ligand traps also have been shown to increase red blood cell production in several clinical trials of patients with chronic anemia .
54.4.4
Acid–base
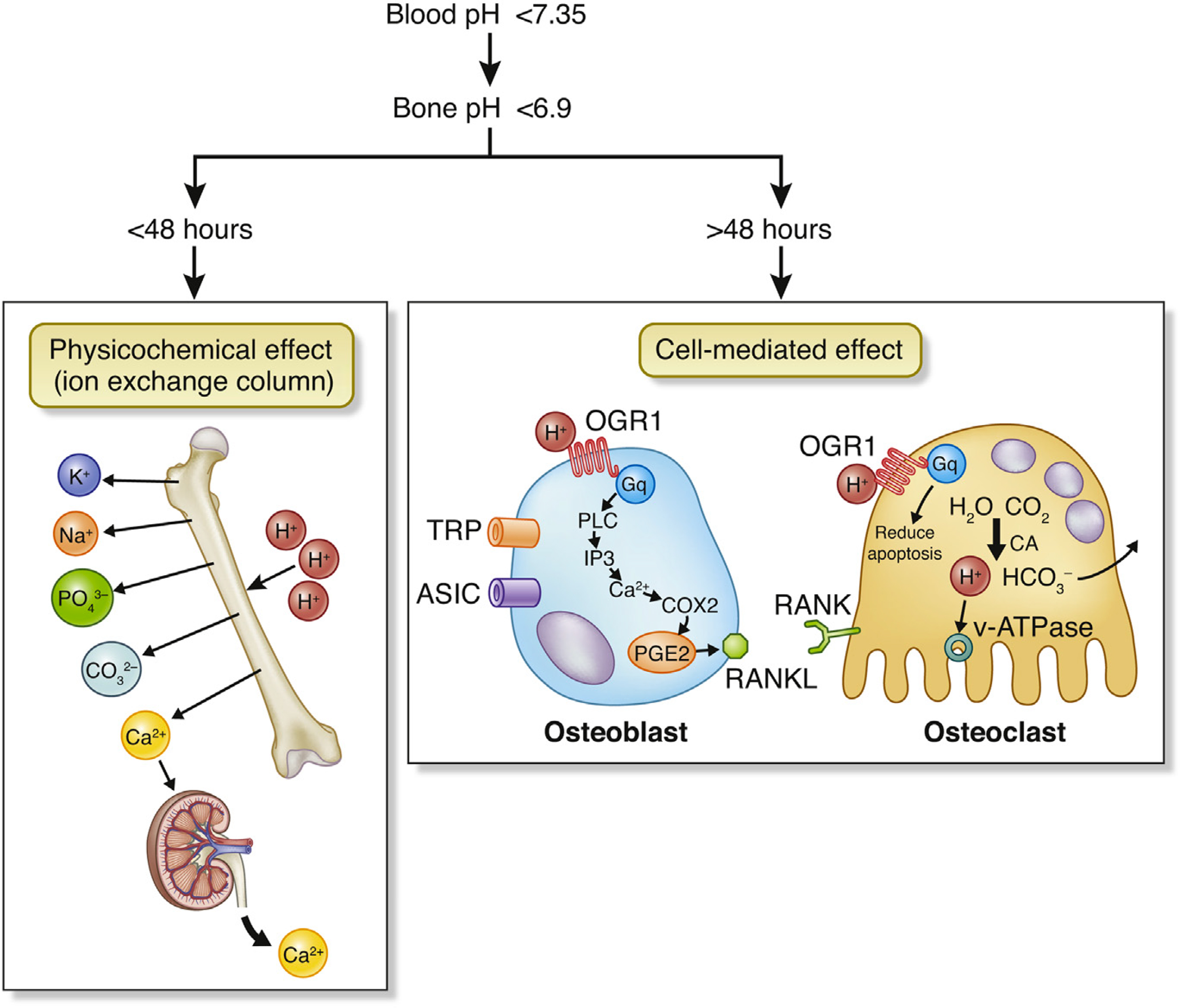
With progressive renal impairment, the kidneys’ ability to synthesize ammonia and to excrete hydrogen ions is reduced, leading to a reduction in bicarbonate levels and metabolic acidosis. In acute metabolic acidosis, calcium and bicarbonate are released from bone in a chemical reaction that does not depend on cellular activity. However, with chronic metabolic acidosis, osteoblast activity is reduced and osteoclastic bone resorption is increased . Both osteoblasts and osteoclasts have a hydrogen ion receptor (OGR1). In the osteoblasts this signals through PLC to increase PGE2 which stimulates release of RANKL, thus increasing osteoclast recruitment. In the osteoclasts, the OGF1 signals pathways that reduce apoptosis . In addition, metabolic acidosis increases osteoblast production of FGF23 ( Fig. 54.7 ).
54.4.5
Abnormal endocrine hormones
54.4.5.1
Gonadal hormones
Levels of gonadal hormones are often abnormal and can contribute to bone disease in later stages of CKD . Premenopausal women with CKD G 4–5 frequently have oligomenorrhea or amenorrhea, and serum estrogen levels are often decreased. However, albumin and sex hormone-binding globulin (SHBG) may also be decreased, so free estradiol levels can be higher than suggested by total estradiol concentrations . From one-third to one-half of men with CKD G3–5 are reported to have disturbances in gonadal hormone levels increasing to 50%–75% of men undergoing dialysis .
Low total testosterone and/or free testosterone levels may result from primary testicular failure or secondary hypothalamic–pituitary dysfunction. Causes include inhibition of luteinizing hormone signaling as a direct consequence of kidney failure , increased prolactin production and reduced renal prolactin clearance , and comorbid conditions such as obesity, diabetes mellitus (DM), hypertension and prescribed medications that can contribute to decreased testosterone levels.
Testosterone undergoes aromatization to estradiol, and adult males have estradiol levels similar to those of premenopausal women during the follicular phase of the menstrual cycle. In fact, estradiol is the hormone that has been most closely associated with regulating bone health in men . In men with CKD G3, estradiol levels are associated with a slight but significant decrease in serum phosphate, although the relationship is stronger in men with normal kidney function . This may be due to an estrogen-induced increase in levels of FGF23, as described in a rodent model . Testosterone also increases serum FGF23 and reduces serum phosphate independent of aromatization to estradiol, although this effect could be indirect due to an increase in levels of serum 1,25D . Postmenopausal women have higher serum phosphate levels than older men or postmenopausal women prescribed estrogen replacement therapy, independent of PTH levels, dietary phosphate intake and eGFR . Responsible mechanisms may include a reduction in estrogen-mediated suppression of bone resorption or reduced proximal tubular sodium-dependent phosphate reabsorption. Interestingly, in women with heart disease and mild-to-moderate CKD, those taking estrogen have both lower phosphate and FGF23 levels than women not taking estrogen .
Circulating testosterone and estradiol are tightly bound to the liver-derived glycoprotein SHBG, which influences the concentration of free hormone available for receptor interactions and biological actions. A recent study of men with end-stage kidney disease awaiting transplantation reported an association of greater age, longer dialysis vintage and lower hip Z -scores to prevalent vertebral fracture, but only higher SHBG levels were associated with prevalent nonvertebral fracture, with no significant association of testosterone or estradiol values to fracture at any site . Similar associations of nonvertebral fracture to higher SHBG values or low bioavailable estradiol have been reported in the Osteoporotic Fractures in Men Study (MrOS) and in The Concord Health and Ageing in Men . Mechanisms remain speculative but include changes resulting from the binding of SHBG to its specific receptor or the influence of SHBG on free sex hormone concentrations.
54.4.5.2
Insulin and osteocalcin
Diabetes mellitus (DM) is the most common cause of end-stage kidney disease in many countries and is a known risk factor for osteoporosis and fractures, particularly involving the lower extremities. For people with type 2 DM the incidence of hip fracture is reported to be two to three-fold that of individuals without diabetes , and in a large population-based cohort study, the hazard ratio for incident hip fracture of participants with type 1 DM, compared to matched participants without diabetes, was 5.64 (95% CI 3.55–8.97) for men aged 60–69 and 5.63 (95% CI 2.25–14.11) for women aged 30–39 . Factors that may contribute to reduced bone quality and skeletal fragility in DM include the accumulation of advanced glycation end products in bone, leading to chemical alteration of collagen, and diabetic microangiopathy . In osteoblasts insulin is anabolic, acting through the osteoblast insulin receptor and stimulating proliferation and differentiation , whereas diabetes suppresses osteoblast function but stimulates osteoclast activity. Hyperglycemia also promotes stem cell differentiation toward adipocytes, increases marrow fat deposition, decreases osteoblast differentiation, and reduces bone formation .
Bone biopsy studies generally report low bone turnover in patients with type 2 diabetes and reduced biochemical markers of bone turnover may reflect these changes. However, glycated peptides such as CTX may not be accurately detected by CTX ELISA assays .
Osteocalcin
Osteocalcin (OC), which has also been referred to as bone gamma-carboxyglutamic acid (Gla) protein, is a small water-soluble vitamin K-dependent protein that is one of the most abundant noncollagenous proteins in bone. It is secreted by osteoblasts in response to vitamin D and PTH and undergoes posttranslational vitamin K-dependent gamma-carboxylation. Within bone, gamma-carboxylated OC is bound to hydroxyapatite and affects crystal size and shape and osteoblast and osteoclast activity. However, uncarboxylated OC (ucOC) is not bound to bone and is released into the circulation together with OC that is released during bone resorption. The serum of people with normal renal function contains from 20% to 50% ucOC and 50% or more carboxylated OC .
Many patients with CKD and on dialysis have low values of vitamin K , which may be due to limitations of dietary phosphate and potassium and a corresponding reduction in foods with high vitamin K content. Vitamin K supplementation of patients on dialysis results in a reduction in circulating ucOC and high dietary vitamin K1 intake, or supplementation with vitamin K2, can increase carboxylated OC .
In patients with normal kidney function, serum OC reflects bone turnover, because levels represent a combination of osteoblast and osteoclast activity. However, OC accumulates as CKD progresses, and in ESKD, serum OC ceases to be a reliable turnover marker. Nevertheless, in patients on dialysis, states of high bone turnover have been associated with increased ucOC levels . This was particularly apparent in patients with high bone turnover assessed by levels of PTH and the osteoclast marker TRAcP-5b . Conversely, patients with diabetes, who tend to have low bone formation, are likely to have lower ucOC levels.
Circulating OC is now known to act systemically to increase insulin sensitivity . In some studies, total, but not ucOC concentrations, have been associated with glycemia, glucose metabolism and measures of adiposity . In others, ucOC and carboxylated OC have been associated with improved glucose tolerance, with the ucOC associated with enhanced β-call function and the carboxylated form with improved insulin levels . However, a study of 189 hemodialysis patients found that higher ucOC correlated inversely with lower plasma glucose, hemoglobin A1C, and glycated albumin . Overall, the active endocrine conformation appears to be ucOC, which stimulates insulin secretion directly, increases intestinal glucagon-like peptide-1 secretion and enhances the release of adiponectin from adipose tissue, thereby increasing the sensitivity of muscle to insulin while carboxylated OC mediates important bone effects. Despite these associations, vitamin K1 supplementation given to postmenopausal women to reduce ucOC did not influence blood sugar levels or insulin resistance suggesting that in humans, the endocrine function of OC requires further investigation.
Malnutrition, insulin-like growth factor
Patients with CKD usually have nutritional problems. Their diets must be restricted because they cannot excrete excess phosphate or potassium or sodium. Nausea and anorexia limit the intake of food. Malnutrition, inflammation, metabolic acidosis, and hormonal factors are all likely to contribute to the development of bone disease and muscle loss.
Circulating insulin-like growth factor 1 (IGF-1) is synthesized in the liver in response to growth hormone and is necessary for kidney growth, and the normal development of lean body mass, longitudinal bone growth, and cortical integrity . Defective IGF signaling activates the degradation of muscle protein and muscle loss. On the other hand, the skeletal synthesis of IGF-1 from osteoblast progenitors and fully differentiated osteoblasts is primarily dependent on PTH. This locally produced IGF-1 stimulates bone formation, stabilizes β-catenin, enhances wnt signaling, and plays a more significant role in the maintenance of cancellous bone integrity . IGF-1 is required to obtain an anabolic response to teriparatide, and the stimulatory effect of abaloparatide, a modified 1–34 amino terminal fragment of PTH-related peptide (PTHrp), is mediated at least in part by enhanced local IGF-1 production . Because it is stored in the bone matrix and released during bone resorption, IGF-1 may also be involved in the coupling of bone formation and resorption.
Serum levels of IGF-1 must be interpreted cautiously, because the bioavailability of IGF-1 is altered in kidney disease . In the US population, serum IGF-1 is positively associated with estimated GFR . However, Jehle et al. studied 319 patients with various grades of kidney disease, including dialysis patients, and found that total and free IGF-1 levels remained constant in most patients. The IGF-1 was positively correlated with the bone formation rate.
Studies report various levels in dialysis patients . The bioavailable levels of IGF may be low even when total levels are increased, because the binding proteins are also increased. Han et al. found lower IGF-1 levels in patients on dialysis than in normal subjects. These patients also had abnormalities in their muscle strength.
54.4.5.3
Bone morphogenetic protein 7
BMP-7 is an important promoter of osteoblast differentiation that is produced primarily by the kidneys, where it is highly expressed and is required for normal kidney development, renal responses to injury, and as a promoter of osteoblast differentiation. BMP-7 also regulates the activity of TGF-β, which is essential for tissue repair and fibrosis but must be tightly controlled. BMP-7 does this by competing with TGF-β for a limited pool of Smad4 . As a result of cell injury, TGF-β expression is upregulated, together with a simultaneous reduction of BMP-7 expression, and as the injured cells recover, BMP-7 expression is restored, to counteract the TGF-β response .
BMP-7 is a growth factor necessary for the differentiation of preosteoblasts into mature osteoblasts, and levels are reduced in CKD . Therefore stimulation of the bone environment by PTH leads to accumulation of preosteoblasts in the marrow, which do not progress to maturity. This histology has traditionally been termed “fibrosis” because the long spindle-shaped cells resembled fibroblasts .
BMP-7 has been used successfully to treat the adynamic bone disorder that developed in adult mice after the induction of renal impairment . Because BMP-7 upregulates the expression of osteoblast PTH receptors, higher levels of PTH may be required for regulation of stem cell niches when levels of BMP-7 are reduced by renal damage .
BMP-7 also lowers expression of some of the genes that have increased expression in CKD, and which are related to arterial wall fibrosis (fibronectin, periostin, activin A, and Snail). BMP-7 can prevent vascular calcification in a rat model, but it does not reduce calcifications once they have occurred .
54.4.6
Accumulation of substances
54.4.6.1
Aluminum
About a decade after long-term dialysis was first used, some centers reported a unique syndrome of dementia. The first symptom was stuttering during dialysis, and this rapidly progressed to seizures, coma, and death within 6 months. The patients also had osteomalacia. The etiology was a mystery and the most likely cause seemed to be a virus. Several centers had epidemics of this disease, while others did not have any cases. Eventually, these outbreaks were linked to the aluminum levels in the water used for the dialysate. Municipal water companies may add aluminum to deflocculate water, and in several cities the dialysis centers started seeing dementia soon after the water department began using this form of water treatment. Meanwhile, investigators had described two forms of osteomalacia in dialysis patients: one responded well to calcitriol but the other was resistant and caused by aluminum . Thereafter, all dialysis centers began using methods to purify the water, and the incidence of aluminum-related osteomalacia dropped dramatically. Currently, only sporadic cases are seen, with high doses of oral aluminum or with accidental contamination of dialysate or parenteral solutions.
54.4.6.2
Iron
Iron accumulation in the bone can also cause osteomalacia in dialysis patients. On bone biopsies, iron deposition is seen along the surface of the bone, particularly at the mineralizing front . This can be seen in patients with frequent blood transfusions. The iron appears to be directly toxic to the osteoblasts .
Iron deficiency results in increased expression of FGF23 (see above section on FGF23).
54.4.6.3
Strontium
Some animal studies suggest that accumulation of strontium may also cause osteomalacia . In rats, the effects are complex and dose-dependent; some had adynamic bone and others developed osteomalacia . Strontium levels are increased in bones of dialysis patients who had osteomalacia . This has not been seen in osteoporotic patients who have been treated with strontium, but they did not have kidney disease . Strontium increases the bone density, partially because the ion is heavier than calcium and replaces calcium in the bone matrix. Also, it inhibits bone resorption. Although it is frequently stated that strontium is anabolic, a large biopsy study documented that strontium decreased the bone formation rate as well as bone resorption rate . There are no clinical trials using strontium in patients with CKD. Use of strontium as a treatment for osteoporosis is no longer recommended, due to an increased risk of myocardial infarction .
54.4.6.4
β2 Microglobulin (amyloid)
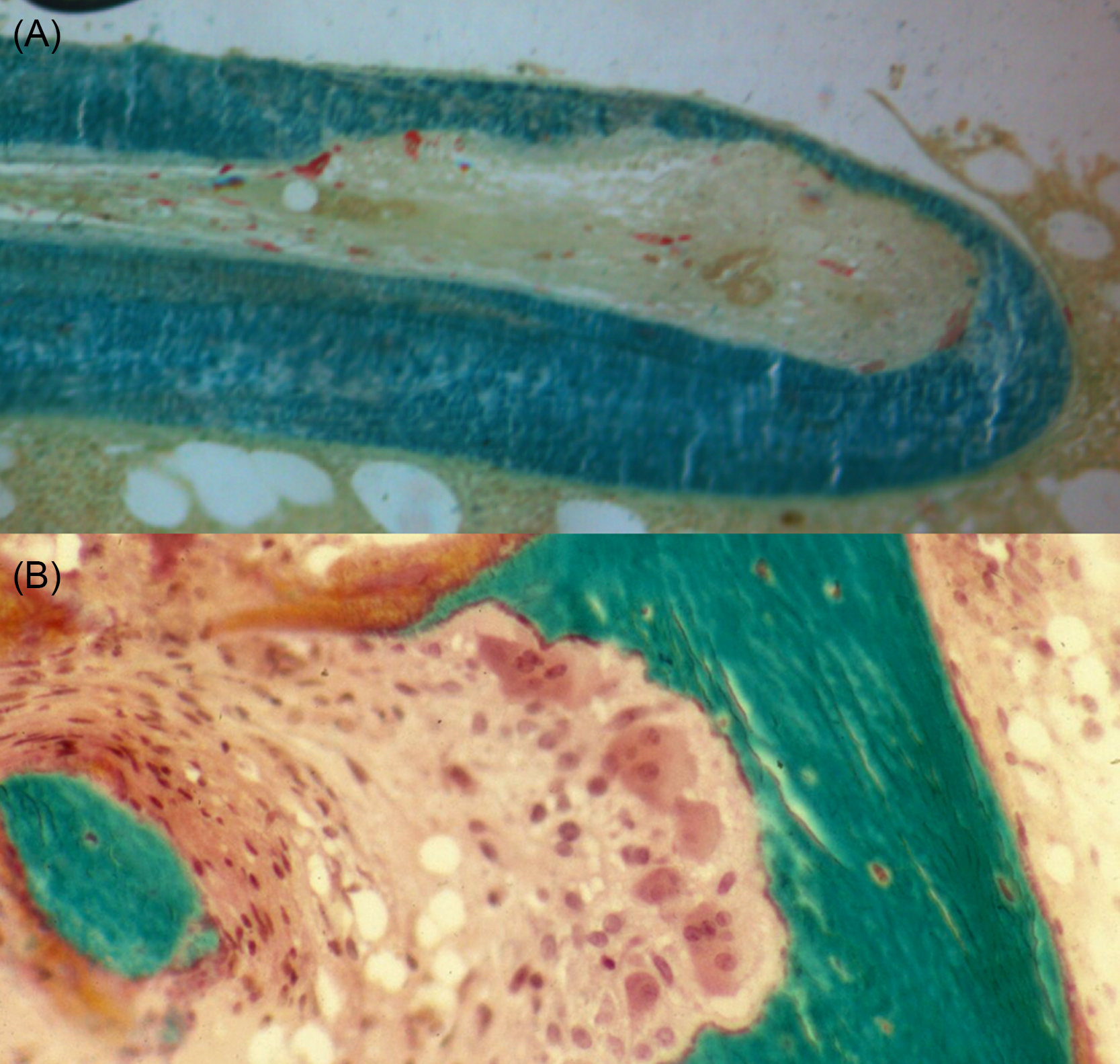
β2 Microglobulin is a byproduct of immunoglobulin metabolism and is normally excreted by the kidney. This protein is too large to be efficiently dialyzed, and it may accumulate in patients after many years of hemodialysis. The protein aggregates form β-pleated sheets, which results in dialysis-related amyloid, but the distribution is different than that of other forms of amyloid. Deposits are seen in the bone near the joints, where it binds to collagen and stimulates osteoclastic bone resorption, forming cystic lesions . Amyloid also deposits around the tendons of the wrist and causes carpal tunnel syndrome ( Fig. 54.8 ) . High-flux dialysis membranes are more efficient at removing the β2 microglobulin .
54.4.7
Low activity and poor muscle strength
Patients with CKD 4–5 have multiple medical problems that may result in decreased physical activity. Measurements of muscle strength by the timed get up and go test, and by the 6-minute walk test, predict fractures in patients with CKD. In one study assessing fracture risk the area under the receiver operating curve with these muscle tests was 0.90, which was higher than the area using high-resolution pQCT at the radius (AUC 0.80) . Muscle strength is just one factor that can lead to falls, and 44% of dialysis patients older than 65 had more than one fall in the previous year . In elderly men and women treated for osteoporosis, CKD is a risk factor for falls . A cohort of 183,047 US patients who initiated dialysis found that over 2 years the cumulative incidence of serious fall injury was 6% . A study of fall prevalence in osteoporotic patients showed a greater risk in those with eGFR less than 65 mL/min/1.73 m 2 , especially when they were taking glucocorticoids .
Patients with CKD 4–5 often have nutritional and metabolic derangements that contribute to a loss of body protein, energy stores, muscle and fat mass, resulting in cachexia, frailty, and decreased physical activity. This state has been termed protein energy wasting (PEW) , and usual diagnostic criteria include weakness, slow gait speed and reduced physical activity, unintentional weight loss, and reduced muscle mass. PEW shares many characteristics of sarcopenia, defined by the 2018 European Working Group on Sarcopenia in Older People by low muscle strength (assessed as grip strength and the Recurrent Chair Stand Test), confirmed by low muscle mass or quality, and graded for severity by tests of physical performance (gait speed, timed-up-and-go test, or the 6-minute walk test) .
Muscle wasting (or sarcopenia) is highly prevalent amongst patients with CKD and may not be simply responsive to protein feeding. In these patients complex mechanisms stimulate loss of skeletal muscle, involving activation of mediators that stimulate the ATP-dependent ubiquitin–proteasome system. Abnormalities in insulin/IGF-1 signaling activate caspase-3, a protease that disrupts the complex structure of muscle proteins . In dialysis patients, grip strength is reduced compared to normal controls and is lower in dialysis patients with higher serum myostatin levels . Relationships between nerve and bone are being discovered, and the neuropathy associated with CKD may play a role in the bone disease. Neuropathy also can be a factor in development of stress fractures of the feet, due to direct effects of nerve signaling to bone and due to reduced proprioception.
Myostatin had emerged as a humoral factor that is a member of the TGF-β family and is produced by skeletal muscle in response to oxidative stress and inflammation. It binds to type II activin receptors, which then activate the type I receptor (ALK) that phosphorylates SMAD 2 and 3, leading to activation of pathways that stimulate protein breakdown and inhibit protein synthesis . In myocytes ALK, activation increases wnt-signaling and transcription of atrophy-related genes. Myostatin also inhibits synthesis of IGF-1. Also, inflammation increases interleukin 6 and upregulates skeletal production of myostatin, helping to explain cachexia in cancer and in CKD .
Given its complex nature, physical interventions and nutritional support may improve but will not reverse the impact of sarcopenia on patients with CKD. A number of pharmacological agents are under investigation for its treatment, including inhibitors of myostatin and inhibitors of RANK ligand, which is expressed in skeletal muscle .
54.5
Skeletal manifestations of chronic kidney disease
54.5.1
Fractures
Fractures are the most serious skeletal complication of CKD. The incidence of fractures is increased, as detailed in the epidemiology section of this chapter.
54.5.1.1
Fractures predict future fractures and mortality
Lower eGFR is associated with both increased fracture risk and postfracture mortality. For elderly patients, levels of eGFR<45 mL/min/1.73 m 2 are associated with a near doubling of hip-fracture related mortality . Among 22,621 patients from a US cohort of patients undergoing hip repair, 377 dialysis-dependent patients compared to nondialysis patients had an odds ratio of in-hospital mortality of 3.13 (CI 1.72–5.7) . Several studies have compared mortality rates between CDK G5D patients with and without fracture, and found rates 1.9–3.7 times greater .
The in-hospital mortality rates after hip fracture in dialysis patients in the United States from 2003 to 2011 have declined by 26.7%. The mortality rate was 8.6% in 2003 and 6.3% in 2011 .
In patients with CKD G 5D, a vertebral fracture identified on a radiograph increased the risk of a new fracture by over sevenfold . The fracture risk assessment tool (FRAX) has also been used in fracture prediction. In a prospective study of 1068 patients on hemodialysis, FRAX (without BMD) predicted major fractures over 2 years, with an AUC of 0.76. A prevalent fracture and glucocorticoid use were the strongest factors in the prediction .
54.5.2
Bone pain
Whereas ordinary osteoporosis does not cause pain until the patient experiences a fracture, renal osteodystrophy is associated with bone pain, especially when there is osteomalacia. Severe secondary hyperparathyroidism may also cause bone pain.
54.5.3
Bone histology
54.5.3.1
Types of renal osteodystrophy
In renal osteodystrophy, bone formation rate varies from unmeasurably low to very high . Superimposed upon this spectrum are variations in the degree of mineralization of the bone matrix and variations in bone volume (BV), which combine to determine the bone density. An international committee sponsored by Kidney Disease: Improving Global Outcomes (KDIGO) concurred that the most important parameters to describe renal osteodystrophy are TMV . Thus instead of a one-dimensional axis, the TMV system defines a more comprehensive three-dimensional space ( Figs. 54.9 and 54.10 ).
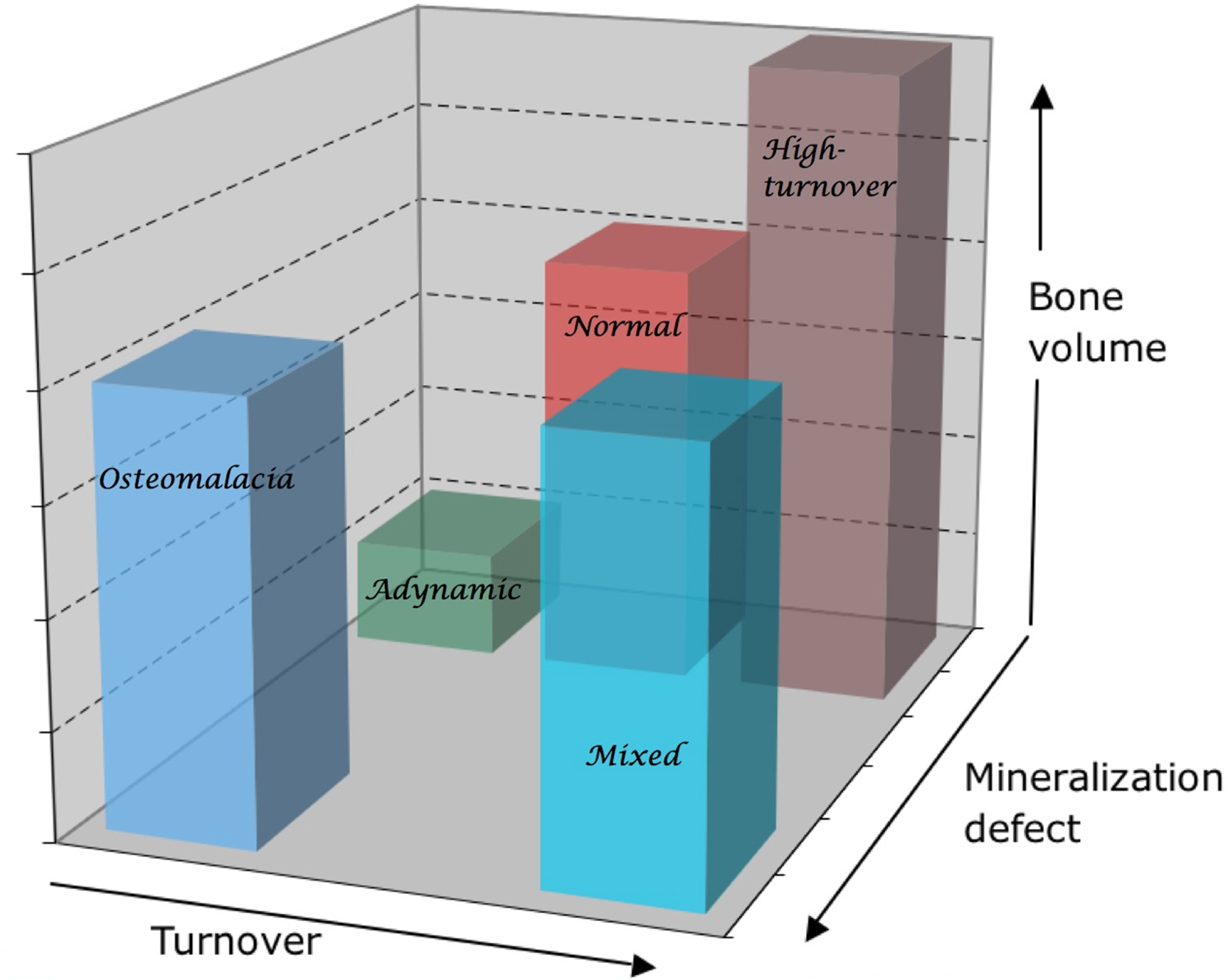
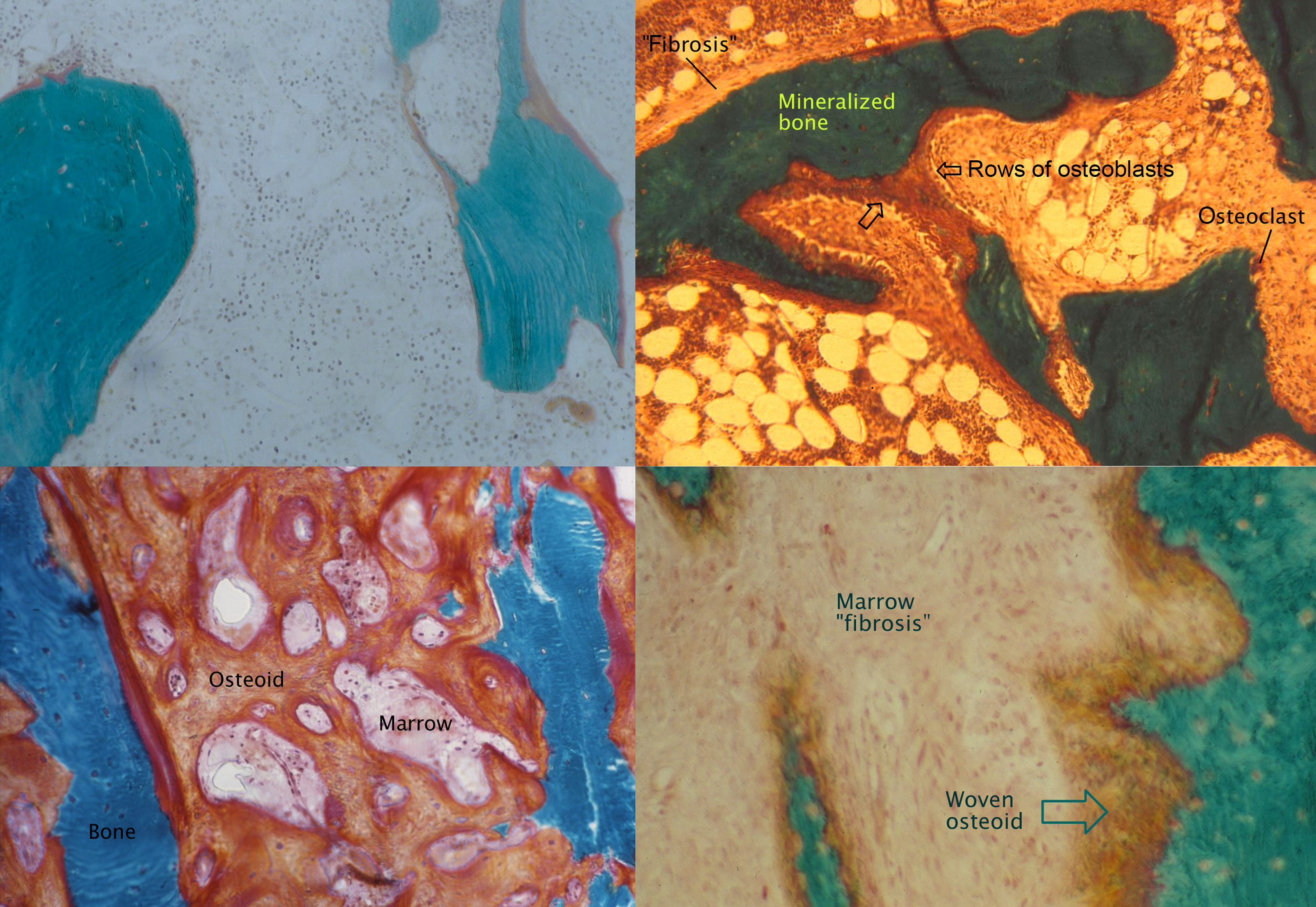
Most older studies of renal osteodystrophy have classified biopsies based on the bone formation rate or presence of fibrosis, as well as the degree of mineralization. The six traditional categories are normal, adynamic (low bone turnover and normal mineralization), osteomalacia (low bone turnover and abnormal mineralization), high turnover, osteitis fibrosa (high bone turnover with “fibrosis”), and mixed (high bone turnover with abnormal mineralization). There is no consensus on the exact measurements to define these categories. A review of 2340 biopsies performed on Brazilian patients for clinical indications found that the frequency of fractures was significantly higher in those with osteomalacia compared with other forms . Another study that followed 62 patients for 5 years after bone biopsy found a higher rate of fractures in those with adynamic bone disease .
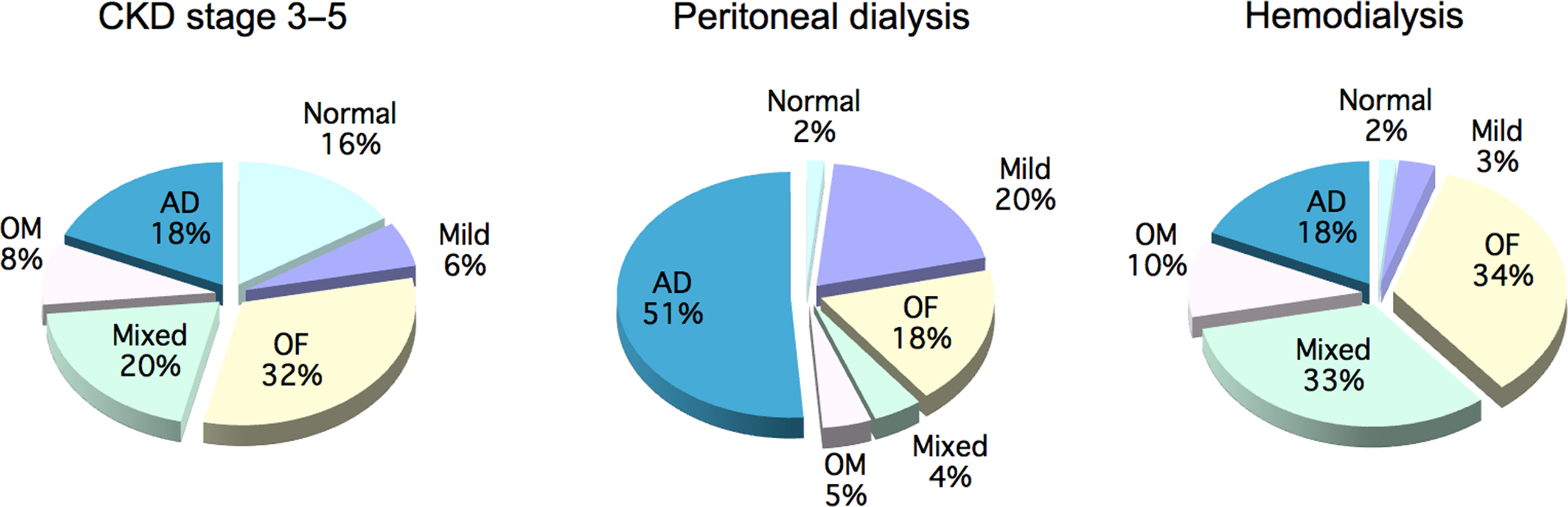
A comprehensive literature review of the prevalence of types of bone disease in CKD is shown in Fig. 54.11 . The review analyzed studies carried out between 1983 and 2006 . The differing prevalence of bone disease types observed between studies is due to differing classification methods, in addition to differences related to geographical areas, genetic background, and treatment modalities ( Fig. 54.11 ).
A report from 2011 shows that the patterns of renal osteodystrophy may be evolving. Malluche analyzed 630 bone biopsies performed in patients with CKD grade 5, from 2003 to 2008, using the TMV system. 58% of patients had low bone turnover, which is a shift from older studies showing a preponderance of high turnover related to hyperparathyroidism. Only 3% of patients had abnormal mineralization. Also, 73% of the white patients with low BV had low bone turnover . It is likely that these differences represent both demographic shifts and different treatment modalities.
The patterns reported from bone biopsies reflect some racial differences. The BV in black dialysis patients is higher than that in whites and the bone formation rate is mostly normal or elevated in blacks but mostly low in whites. This is in contrast to black patients with normal renal function who have low bone formation. In the dialysis population the black patients had higher PTH levels than white patients (661 vs 274 pg/mL), but at any level of PTH, bone turnover was lower in blacks than in whites, indicating resistance to the resorptive effects of PTH. The black patients were treated more often with vitamin D or its metabolites. Also, black dialysis patients have a greater survival rate than white patients .
54.5.4
Bone biochemical markers
54.5.4.1
Relationship of parathyroid hormone to fractures and mortality
In patients with CKD grade 5D, several large studies relating serum PTH to fractures have provided inconsistent results, possibly because both high and low levels are harmful ( Table 54.6 ).
| Author | Year | N | Fractures |
|---|---|---|---|
| Coco | 2000 | 1272 | High risk with low PTH |
| Stehman-Breen | 2000 | 4952 | No relation |
| Block | 2004 | 40,538 | Weak direct association, P =.035 |
| Danese | 2006 | 9007 | Higher risk with low or high PTH |
| Jadoul | 2006 | 12,782 | Relative risk=1.7 if PTH>900 |
| Mitterbauer | 2007 | 1774 | No relation |
| Iimori | 2012 | 485 | Higher risk with low or high PTH |
| Urena | 2003 | 70 | No relation |
| Negri | 2004 | 65 | No relation |
| Inaba | 2005 | 114 | No relation |
| Elder | 2006 | 242 | More after PTX otherwise not related |
| Ersoy | 2006 | 292 | No relation |
| Rudser | 2007 | 5918 | Fewer after PTX |
| Jamal | 2006 | 52 | No relation |
| Atsumi | 1999 | 187 | More with low PTH |
The PTH level can impact the ability of bone density to predict fractures. In a prospective cohort study of 485 Japanese hemodialysis patients with a mean age of 60 years, femoral neck BMD did not predict fracture when the PTH values were above the median(204 pg/mL) but showed reasonable predictive capacity when PTH was lower than the median (AUC of 0.717) . A case-controlled cohort study found a 31% (95% CI 0.57–0.83, P <.001) reduction in global fracture risk following parathyroidectomy .
Evaluation of mortality risk in 413 incident dialysis patients in Japan showed that lower PTH (<65 pg/mL) was independently associated with mortality [HR 2.06 (95% CI 1.11–3.83)] as well as major cardiovascular events. The low PTH group also had greater baseline aortic calcifications and were older, had more diabetes, higher CRP levels, and lower albumin, but these were included as covariates in the analysis .
54.5.4.2
Association of other markers of bone turnover to fractures
An association between high serum ALP levels and the risk of fractures was reported in 2008 , and in a later, prospective study , higher serum values of bone-specific alkaline phosphate (bALP) predicted incident fractures. Similarly, a positive association between higher serum ALP values and fracture risk was reported from a nation-wide study of 185,277 Japanese hemodialysis patients (mean age 66±12 years and median dialysis duration 5.8 years) . During 1-year follow-up, incident hip fracture occurred in 1586 patients (1.0%), and in multivariate analysis, patients in the highest ALP quartile had higher all-cause and cardiovascular mortality, and a higher incidence of hip fracture than those in the lowest quartile (HR 1.71, 95% CI 1.33–2.18). When combined with PTH levels, the highest mortality and fracture rates were seen with highest ALP and lowest PTH .
In cohorts of elderly women, most of whom have early grades of CKD, serum biochemical markers of bone turnover have been associated with fractures. However, for patients on dialysis there are limited data. Urena et al. found that serum type I collagen cross-linked telopeptide (CTX) and bALP were not different between fracture and nonfracture cases in a survey of 70 dialysis patients. Aleksova et al. reported that the bone turnover markers procollagen type I N-terminal propeptide (P1NP) and CTX were positively associated with femoral neck cortical thickness assessed using DXA but were not associated with prevalent fracture.
Another study evaluated CKD patients without known cardiovascular disease and found that reduced Tartrate-resistant acid phosphatase (TRAP) and elevated bALP were both associated with an increase in the relative risk of cardiovascular mortality . These somewhat paradoxical findings suggest that much more work needs to be done to fully understand the clinical utility of such biomarkers.
54.5.4.3
Parathyroid hormone and turnover markers in chronic kidney disease in relation to bone histology
Hyperparathyroidism continues to be a major cause of renal osteodystrophy. The classic findings of renal hyperparathyroidism are increased numbers of multinucleated osteoclasts, woven bone, blurry tetracycline labels, increased cancellous BV but decreased cortical thickness, and intratrabecular tunneling ( Fig. 54.12 ). Osteitis fibrosa is in fact a misnomer; there is no active inflammation and the “fibrosis” is predominantly cellular with some extracellular matrix, which can disappear rapidly when PTH is lowered. The cells are not fibroblasts but inchoate preosteoblasts that proliferated under the influence of PTH but did not achieve mature differentiation. Their differentiation to mature osteoblasts can be corrected by addition of BMP-7 . However, the bone response to PTH is not consistent, and there is evidence for skeletal resistance to PTH in patients with CKD-MBD. This is why guidelines recommend that serum levels of PTH should be higher than the range seen in normal individuals .