48.1
Scope of the problem
Although osteoporosis is primarily considered an issue for postmenopausal women and men over age 65 years, physicians are increasingly confronted with low bone mass or fractures affecting a younger population . This is facilitated by three factors: (1) the increased awareness of osteoporosis as a cause of fractures in younger individuals; (2) the availability of bone density measurements, and (3) the availability of different treatment options for osteoporosis in younger individuals. Because osteoporosis is recognized with increasing frequency in young adults, the clinician is faced with a differential diagnosis that may include an inherited disorder such as osteogenesis imperfecta (OI) or acquired endocrine, gastrointestinal, and renal disorders. These include hyperparathyroidism, Cushing syndrome, occult malabsorption (e.g., celiac disease), and idiopathic hypercalcemia. Osteoporosis due to occult malignancy must also be considered in a young adult. A difficult therapeutic decision confronts the clinician when secondary causes of osteoporosis are excluded, and the remaining diagnosis is primary osteoporosis in a young adult .
The diagnosis of primary osteoporosis in young adults between the ages of puberty and 50 years invokes two issues: (1) is there an identifiable genetic factor influencing bone mass and (2) how does the clinician define the role of a genetic disorder in relation to low bone mass? Multiple factors may underlie a failure to achieve peak bone mass during the teenage years which will lead to osteoporosis in later life . Osteoporosis related to a heritable disorder may first be recognized in infants or not until adulthood. Osteopenic young adults (<50 years) presenting with a first time nontraumatic fracture are encountered in clinical practice. Our experience indicates that the significance of fragility fractures in young adults may be overlooked.
This chapter addresses the differential diagnosis of osteoporosis in young adults, in particular where the clinical findings suggest OI, idiopathic osteoporosis (IOP), or other genetic disorders that have clinical features in common with these syndromes.
48.2
Osteogenesis imperfecta as a cause of adult osteoporosis
48.2.1
Introduction
OI (OMIM 166,200; 166,210; 166,220; and 259,420), the “brittle bone” syndrome, was listed among the heritable disorders of connective tissue by Victor McKusick in 1972 . Interest in this disorder has been promoted by two developments: first was the application of bisphosphonate treatment to decrease fracture risk and second was the definition of several genes now recognized as responsible for recessive forms of OI. However, as of this writing, it is clear that (1) a successful treatment that uniformly decreases fracture incidence has yet to be defined for children and adults and (2) future research will discover yet additional mutations responsible for the OI phenotypes.
48.2.2
Definition
OI is an inherited systemic disorder of connective tissue characterized by fragile bones (fragilitas ossium), blue sclerae in many individuals, dentinogenesis imperfecta (DI), hearing loss, and short stature. OI is a disorder of type I collagen synthesis. It is now apparent that mutations causing OI may involve the synthesis of procollagen α-1 or α-2 chains, as well as multiple enzymes involved in the normal intracellular processing of type I collagen . The phenotype of OI is extremely varied. Mildly affected individuals may have few fractures, normal height, and near-normal bone density. Severe OI is associated with multiple fractures and skeletal deformities that require wheelchair dependence. OI types II and VIII may be lethal in the perinatal period.
48.2.3
Clinical classification of osteogenesis imperfecta
The classification of OI types I–IV was initially proposed by Sillence in 1979 and modified in 1986 at the seventh International Congress of Human Genetics, Berlin . In 2010 a new revision of the nosology and classification of genetic skeletal disorders was proposed. It was recommended that the Sillence classification be retained as the prototype for classifying severity, and because of phenotype variability, that the several genes involved OI be listed separately to minimize a potentially confusing proliferation of OI types based solely on a specific gene mutation .
The four Sillence types were found to contain type I collagen mutations and to be inherited as dominant traits. Four additional phenotypes, termed OI types V, VI, VII, and VIII, have been designated based on both clinical and genetic characteristics ( Table 48.1 ). Accurate clinical designation is difficult in as many as 25% of OI individuals because of overlap in clinical features between types. To illustrate the confusion caused by having as many related phenotypes and multiple causative genes, Sillence type II OI is lethal and caused by autosomal-dominant type I collagen mutations. Recently designated OI type VIII is severe or lethal but is a recessive trait that is associated with mutations in the gene complex CRTAP-LEPRE1-PPIB ( Table 48.1 ). Thus Van Dijk et al. have proposed a revised classification of OI with exclusion of OI types VII and VIII since these types have been added because of genetic criteria (autosomal-recessive inheritance) while the clinical and radiological features are indistinguishable from OI types II, III, and IV .
| OI type | Phenotype | Inheritance | Biochemical |
|---|---|---|---|
| I | Mild | AD | Null mutation due to premature stop codon COL1A1, COL1A2: normal collagen but half normal amount |
| Blue sclera | |||
| Little bone deformity | |||
| II | Lethal, beaded ribs broad or narrow long bones, thin calvarium pulmonary insufficiency | AD | Structural alteration in COL1A1, COL1A2 |
| III | Progressively deforming, DI, short stature, scoliosis, wheelchair dependent | New mutation, AD | Structural alteration in COL1A1, COL1A2 |
| IV | Moderate skeletal deformity, sclera lighten with age, scoliosis, DI | AD | Structural mutation in COL1A1 and COL1A2 |
| V | Variable phenotype mild to severe | AD | No mutation defined |
| White sclera, dislocation radial head, interosseous membrane calcification, hyperplastic callus, no DI | |||
| VI | Moderate severity, white sclera. Early-onset fractures, osteomalacia on bone biopsy | Not determined | SERPINF1 |
| VII | First Nations Quebec families, moderate-to-severe, rhizomelia, no DI | AR | CRTAP, LEPRE1 |
| VIII | South African black people | AR | LEPRE-1 mutation |
| Severe or lethal |
To illustrate the problem related to phenotypic overlap, type I (mild) OI is uniformly associated with blue sclerae. Scleral color is usually white in adults with type III disease but it may occasionally be blue in children and in some type III adults. Type IV OI is defined on the basis of sclerae that are blue at younger ages but white in adults . However, some adults considered as type IV (moderately severe) also have blue sclerae. Furthermore, blue sclerae occur in several of the heritable disorders of connective tissue [e.g., in some patients with Ehlers–Danlos syndrome (EDS)] as well as in otherwise normal individuals. Because of phenotypic variability, strict assignment of a clinical type early in the course of OI should be avoided because the long-term prognostic value of such assignment is limited, particularly with respect to predicting fracture incidence or the future level of disability, or the eventual social and occupational achievements of the individual.
The definition of OI type is also complicated by the fact that, at this time, there is no firm correlation between a clinical phenotype and a genotype as related to a specific type I collagen mutation (see 48.4.1.1 –Mutations).
48.2.4
Prevalence of osteogenesis imperfecta
OI has been reported throughout the world with no selection for race or gender. It is estimated that 0.008% of the world’s population have OI so that about 500,000 people worldwide have OI There are probably 25,000 affected individuals in the United States and an equal number unrecognized because of the mild nature of their disorder. Approximately 3900 people have OI in the United Kingdom. The estimate of occurrence for severe or lethal disease is about 3 or 4 cases/100,000 births. In Northern Ireland the minimum prevalence rate of OI type II is 0.15/10,000 live births . Less severe disease is estimated to occur in 4 or 5 cases/100,000 births . Estimates based on the presence of fractures at birth range from 1.6/100,000 births in Singapore to 3.3/100,000 in France and 15/100,000 in the United Kingdom . The incidence of OI is also underestimated in the case of aborted lethal or severe type III cases where diagnosis at birth is not possible. Familial transmission of OI occurs in approximately 60% of cases. The estimated incidence of sporadic, as opposed to familial, disease ranges from 19% to 34% . Recessively inherited forms account for approximately 3%–5% of cases of OI in North America and Europe. The incidence of OI based on parental mosaicism is not known, but approximately 6% of familial recurrent cases in the United States may result from gonadal mosaicism.
48.3
Type I collagen and osteogenesis imperfecta
Type I collagen is the major structural protein of bone, skin, tendon, ligaments, and dentin. Type I collagen is also a structural protein for blood vessels, heart valves, and the aorta.
The collagens are a family of proteins that share certain structural similarities but exhibit diverse functional properties. Collagens are formed by three polypeptide a chains assembled in a triple-helical configuration ( Fig. 48.1 ). Each pro-α chain contains a signal region and pro- and telopeptides domains. With respect to α-chain composition, collagens may be homo- or heterotrimeric. Triple-helical domains have a high content of amino acids, glycine, and charged amino acids. At least 28 collagen types the products of 43 genes have been identified . The collagen genes are large: COL1A1 (chromosome 17q23.3–q22) contains, 51 exons and COL1A2 (chromosome 7q21.3–q22) contain 52 exons.
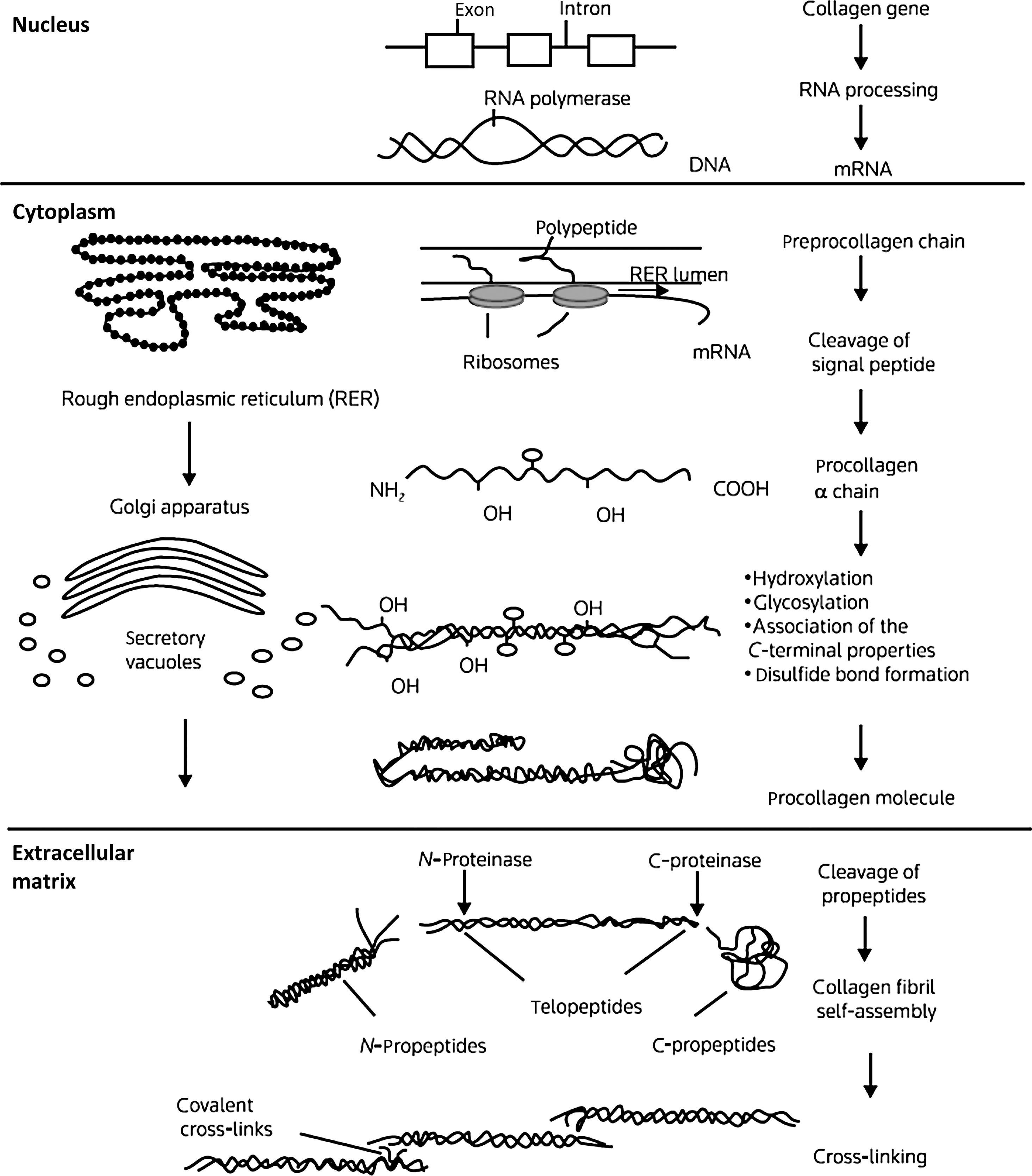
The basic unit of the collagen (I) chain is the repeating triplet (Gly-X-Y) 338 . Approximately 20% of the X and residues are proline which in the Y position is hydroxylated to 4-hydroxyproline. α 1 (I) Proline-986 is hydroxylated at the three position. Glycine residues facilitate the helical configuration due to their small size and repeating position in the triplet. Collagen biosynthesis involves a series of complex intracellular posttranslational modifications ( Fig. 48.1 ) . Hydroxylation of proline residues stabilizes the triple-helical configuration while the formation of lysine aldehyde groups (lysyl oxidase mediated) facilitates the formation of intramolecular cross-links. The collagen-chaperone proteins, SERPINH1 (HSP47) and SERPINF1, which interact with Gly-X-Y repeat in the triple-helical region to stabilize the procollagen molecule in the endoplasmic reticulum . As discussed later, mutations affecting several chaperone proteins which stabilize collagen processing in the rough endoplasmic reticulum have now been identified in severe cases of OI.
Intracellular procollagen processing and its subsequent secretion into the extracellular space leads to cleavage of N- and C-procollagen extension peptides by specific N- and C-terminal proteases. The protein bone morphogenetic protein (BMP)-1, a metalloproteinase, has now been identified as one of the three C-proteinase isoenzymes . Self-assembly and cross-linking of individual triple-helical molecules into a large collagen polymer occurs in the extracellular space. Hydroxylation and glycosylation (glycosyl transferases) of lysine residues permits the formation of stabilizing cross-links between collagen fibrils. The cleaved C-terminal propeptide extensions are not further metabolized but circulate in plasma. Measurement of N- and C-propeptides is used clinically to estimate collagen resorption .
Collagens have been separated into several groups: class I collagens are fibril-forming collagens (types I, II, III, V, XI, XXIV, and XXVII) that include other collagen types incorporated in each fibril. Class II collagens comprise collagen types IX and XII, which adhere to the surface of banded (class I) collagens. Class III collagens include molecules that form independent fiber systems, such as basement membranes (type IV), beaded filaments (type VI), and anchoring fibrils (type VII), as well as type X collagen, which forms a network surrounding hypertrophic chondrocytes in cartilage and at the growth plate. Class IV collagens contain several proteins with unknown fiber forms and with undefined functions. The “FACIT” or fibril-associated collagens with interrupted triple helices are types IX, XII, XIV, XVI, XIX, XX, XXI, XXII, and XXVI. These collagens have short triple-helical regions interrupted by short noncollagenous segments. Transmembrane collagens, including collagens XIII, XVII, and XXV have a short cytosolic N-terminal domain and long triple-helical extracellular domains . Collagen proteins exhibit unique functional specificity. This is illustrated by the large type I triple-helical collagen polymers that provide strength and elasticity to bone matrix and tendon, the short type VII fibrils that form anchoring fibrils, which bind epithelial membranes to dermis, and the type IV and VIII collagens that form basement membranes and Descemet’s membrane . In addition to types IX and XII collagen, types III and V collagen are associated with the surface of type I collagen and that type XI is associated with type II collagen . Illustrative of the complexity of the matrix environment for collagen is the complex role of proteoglycans in fibril formation and collagen stabilization. Members of the small leucine-rich proteoglycans family, decorin, biglycan, fibromodulin, and lumican, bind to collagen fibrils and modulate collagen fibril formation . Two large modular proteoglycans, aggrecan and hyaluronan, enhance the capacity of structural tissues such as tendon to counter the mechanical forces associated with loading and mobilization .
48.4
Clinical overview of osteogenesis imperfecta
Approximately 60% of recognized OI cases are type I, an estimated 10% are type II, 20% are type III, and 5% are classified as type IV. These OI types, initially classified by Sillence, are inherited as dominant traits . The incidence of type V OI is undocumented. The recessively inherited OI types VI, VII, and VIII, account for approximately 3%–5% of the OI population.
48.4.1
Type I osteogenesis imperfecta
Type I OI is the least severe and the most prevalent form of the disease ( Fig. 48.2 ). However, even within this group, there is considerable clinical heterogeneity. Transmission is autosomal dominant. The diagnosis may be missed in very mild cases even with several affected members in one family . For example, a 52-year-old woman considered to represent a case of postmenopausal osteoporosis was reported in whom an α-2 (I) glycine 661→serine collagen mutation consistent with OI was subsequently found . However, attention to the history and physical findings revealed that her first of five fractures occurred at age 7 years and that her 26-year-old son had also suffered fractures. The patient had blue sclerae and slight hearing loss. Thus, although the age of onset or number of fractures may be of little help in establishing the diagnosis of OI, the family history may also be important in establishing the probable genetic basis of the disorder.
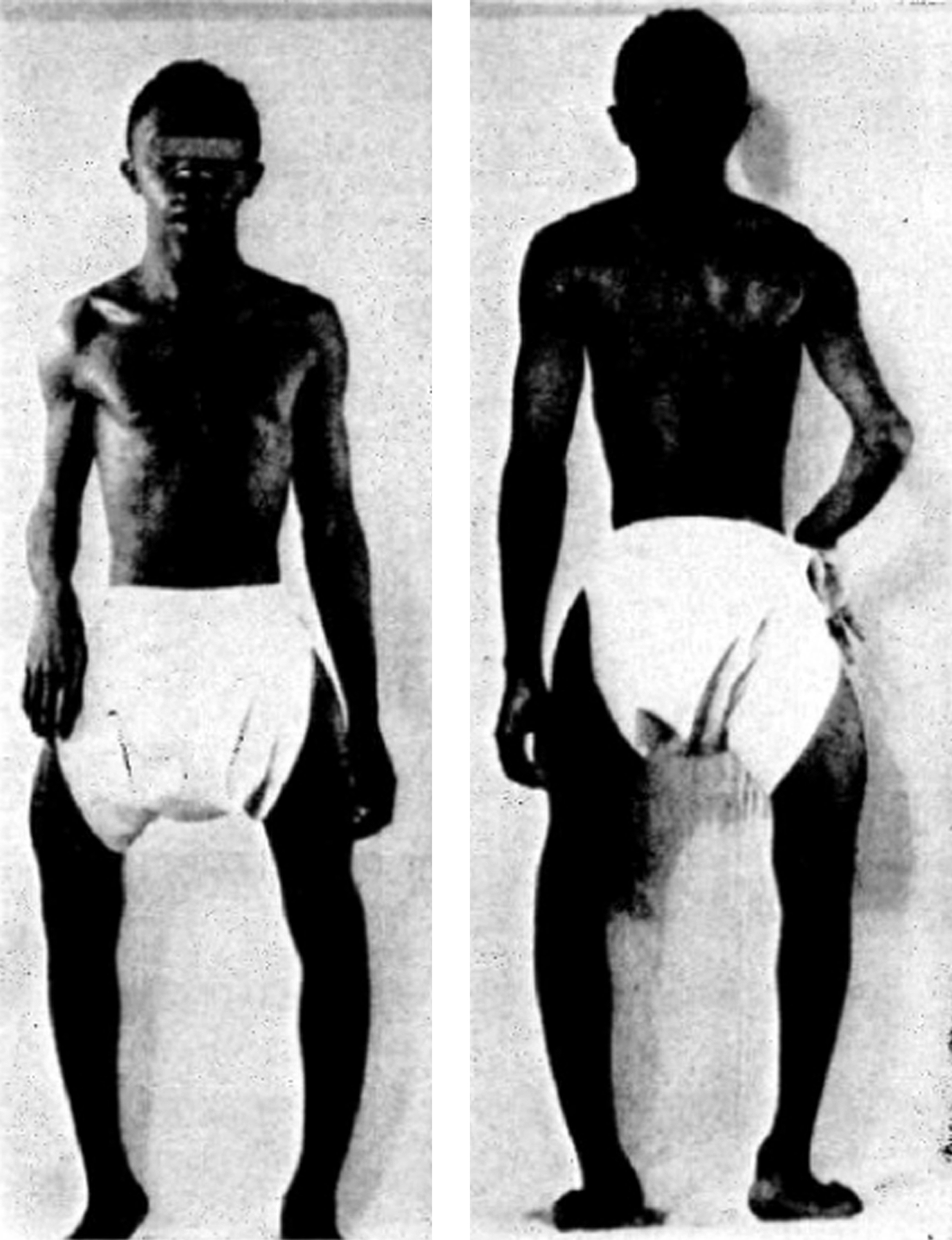
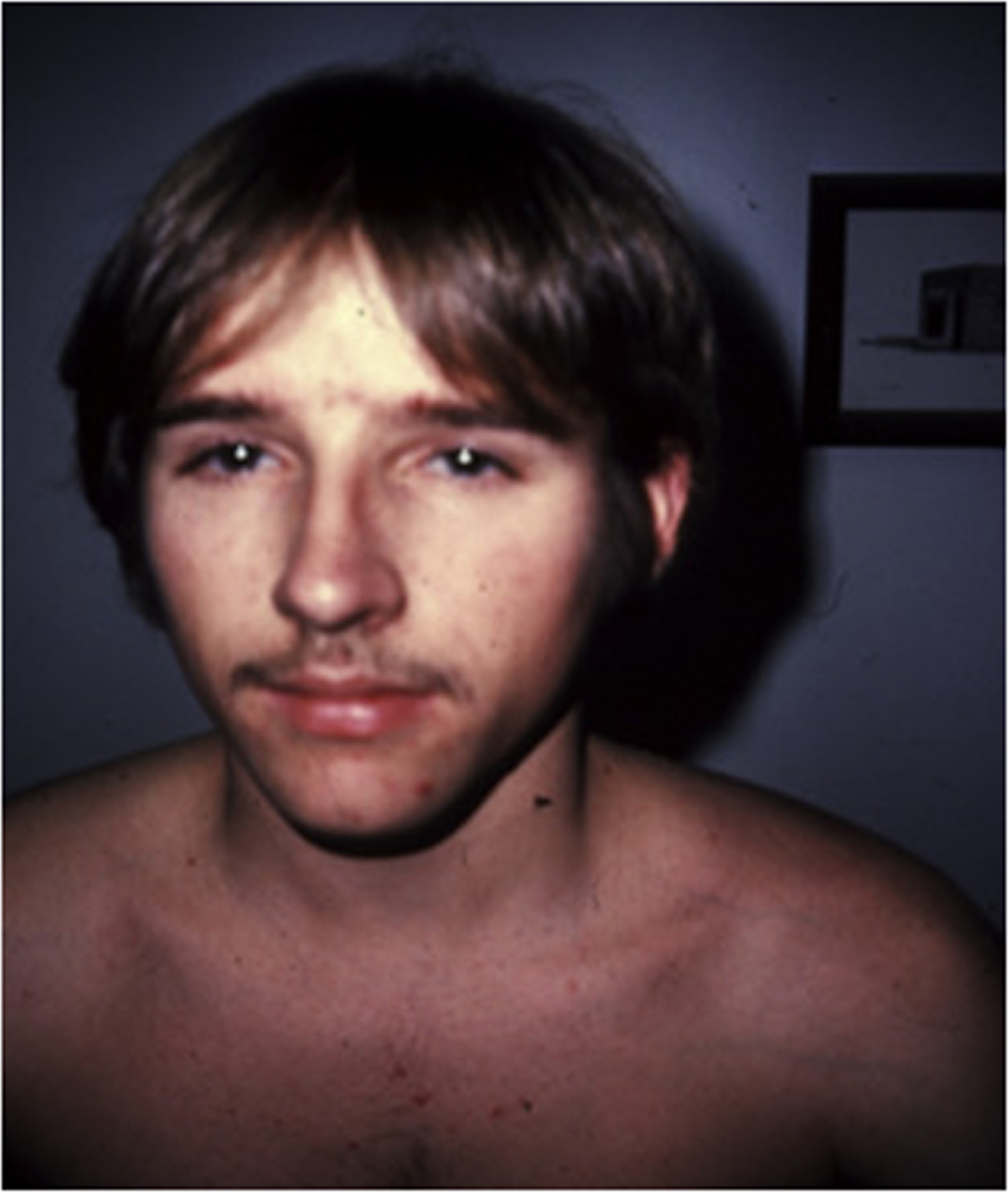
The cardinal manifestations of type I disease include a history of multiple fractures, usually dating from birth or childhood, blue sclerae, hearing loss that is evident between ages 20 and 30 years, mild joint laxity, and short stature ( Fig. 48.3 ). One-third of type IA patients (A=no DI) is less than the 3rd percentile in height, an equal number are between the 3rd and the 50th percentile, and 10% may have normal height. Subjects with type IB (B: with DI) OI tend to be shorter . Although occasional type I individuals have normal stature they will be shorter than their family’s mean height. A characteristic triangular facies due to modeling of the mandible occurs in many patients ( Fig. 48.4 ). In spite of multiple fractures, and in contrast to more severe OI types, skeletal deformity may be mild or absent. DI occurs in approximately 20% of OI subjects . The fracture rate is reported to be higher in subjects with DI . Individuals with type I disease may have fractures at birth, but usually have fractures in early childhood: they may not experience a first fracture until their teens or later. Fracture incidence has a biphasic pattern, decreasing after puberty, and rising again in women and men with increasing age . Scoliosis is also of a mild extent, approximating 15–30 degrees.
Radiologic examination shows a well-proportioned outline of the appendicular skeleton with intact epiphyseal architecture ( Fig. 48.5 ). There are varying degrees of osteopenia that may approach normal bone density in certain patients . Vertebral osteoporosis is usually present and vertebral compression fractures may occur in young adults.
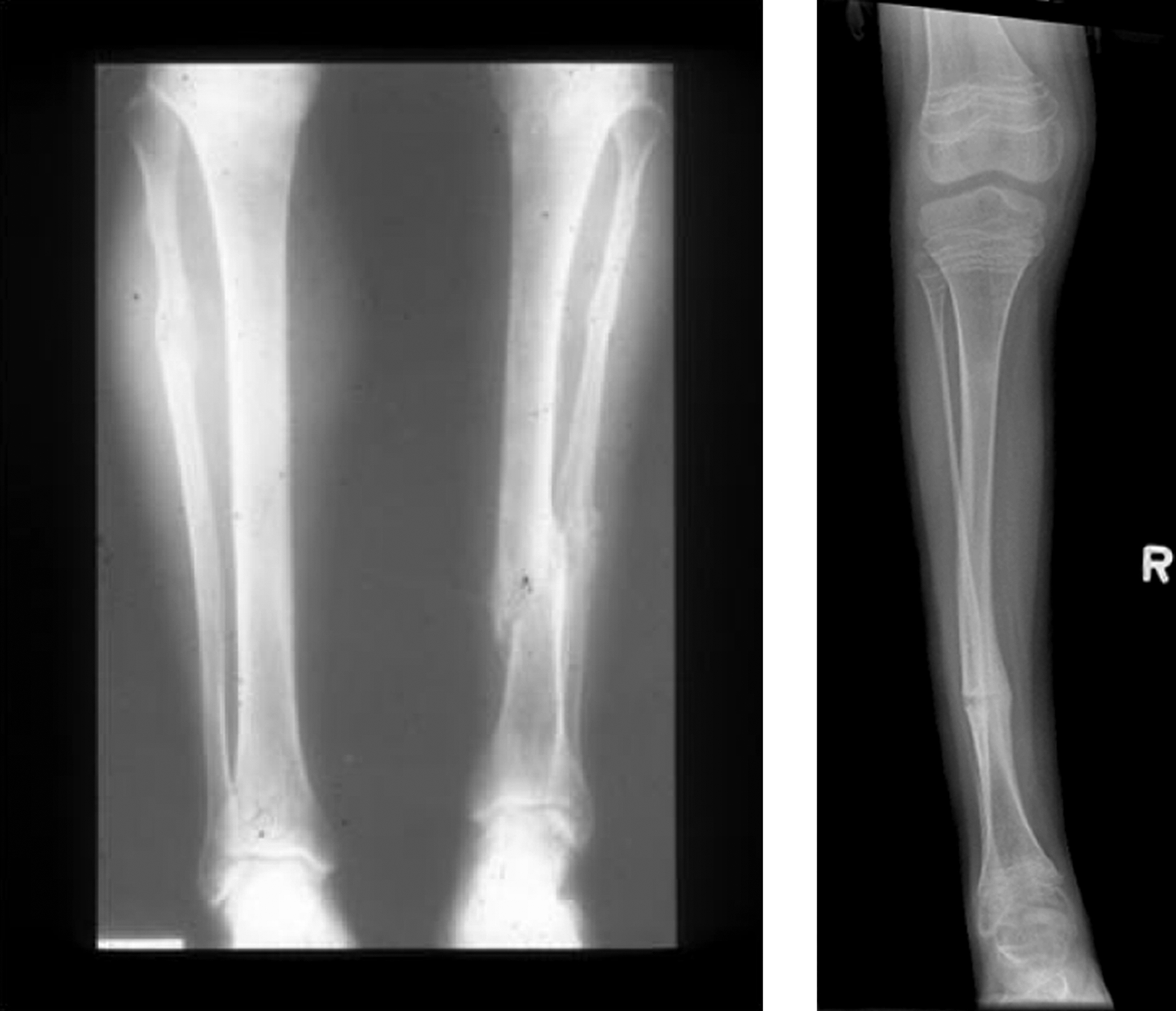
However, there are a small number of mild type I OI cases, in whom mutations in type I collagen a chains have been found who have not had fractures despite having radiologic osteoporosis, blue sclerae, joint laxity, and short stature. Here, the differential diagnosis would include IOP in a young adult (vide infra). However, distinctions have been drawn between these syndromes. In the author’s experience, subjects with IOP have white sclerae, do not have DI, and tend to be taller than individuals with type I OI. In common with OI, these individuals have mild joint laxity, mild scoliosis and may have pectus excavatum. Mitral valve prolapse may be present.
Examination of tetracycline-labeled iliac crest bone biopsy specimens from adults with type I disease have revealed low bone turnover, rather than the high remodeling rate previously reported for children or other OI types .
48.4.1.1
The genetics of type I collagen in osteogenesis imperfecta
Previously, mapping mutations in the large type I collagen genes posed a technical obstacle. Several methods were used to define collagen mutations in fibroblasts derived from skin biopsies and from cultured osteoblasts. Dermal fibroblast cultures are infrequently used now except when uncertainty remains after direct DNA sequencing of collagen genes and confirmation by protein gel electrophoresis using cultured fibroblasts is indicated. This is informative in only a very small number of cases.
Molecular diagnosis is currently largely based on the Sanger method for DNA sequencing which is well known for the accuracy of sequencing it provides . The Sanger method has been considered to represent the gold standard for screening variants of genes. However, Sanger sequencing, at most, can sequence only a small number of exons in one gene at a time. Exome sequencing—a technique which focuses on only the protein-coding portion of the genome—provides many advantages compared to the more expensive whole genome sequence technology .
The dominantly inherited OI phenotypes (OI phenotypes I–IV) are the result of mutations involving the COL1A1 and COL1A2 genes. Table 48.2 lists genes in recessively inherited OI whose products modify procollagen a chains during intracellular processing: CRTAP, FKBP10, LEPRE1, PLOD2, PPIB, SERPINF1, SERPINH1, SP-7, and BMP-1 .
| COL1A1 | Collagen, type I, α1 |
| COL1A2 | Collagen, type I, α2 |
| CRTAP | Cartilage associated protein |
| FKBP10 | FK506 binding protein 10, 65 kDa |
| LEPRE1 | Leucine proline-enriched proteoglycan (leprecan) 1 |
| PLOD2 | Procollagen-lysine, 2-oxoglutarate 5-dioxygenase 2 |
| PPIB | Peptidylprolyl isomerase B (cyclophilin B) |
| SERPINF1 | Serpin peptidase inhibitor, clade F (α-2 antiplasmin, pigment epithelium–derived factor), member 1 |
| SERPINH1 | Serpin peptidase inhibitor, clade H (heat shock protein 47), member 1 (collagen-binding protein 1) |
| BMP1 | Bone morphogenetic protein-1 |
| SP7/Psterix | Sp7 transcription factor |
The reader can access a current list of these mutations in the Human Type I and Type III Collagen Mutation Database [OI and EDS variant databases compiled by R. Dalgliesh ( http://www.le.ac.ul/genetics/collagen )] .
48.4.1.2
The concept of dominant/negative mutations
Dominant/negative mutations involve an altered gene product that acts antagonistically to the wild-type allele. Type I procollagen is a heterotrimer consisting of two identical pro-α 1 (I) chains and a structurally different pro-α 2 (I) chain. A mutation affecting one α 1 (I) allele will alter the synthesis of 50% of those chains with incorporation of either one or two mutated chains into three-quarters of the total number of procollagen molecules. Thus the negative effect of the one dominant mutation is amplified. With the α 2 (I) chain a mutated pro-α 2 (I) would be incorporated into 50% of the type I molecules. This assumes equal production of the mutated and normal chains as well as equal access to procollagen assembly. This process is applicable to the severe or lethal OI types where structurally abnormal proa-chains are secreted into the bone matrix.
48.4.1.3
COL1A1 and COL1A2 mutations in type I osteogenesis imperfecta
Collagen mutations have been classified according to the locus of the mutation . These include (1) helical domain mutations (glycine substitutions), splicing mutations, and helical deletions and insertions, (2) C-telopeptide and C-propeptide mutations, and (3) N-telopeptide and N-propeptide mutations. Mutations are also described as (1) quantitative (e.g., null allele or haploinsufficiency-related mutations) or (2) as qualitative mutations where mutated pro-α chains are assembled in the endoplasmic reticulum and the trimeric molecule is secreted in the extracellular matrix (ECM). The qualitative mechanism operates in OI types II, III, and IV which are associated with either lethal disease or a more severe phenotype.
The most frequently reported COL1A1 and COL1A2 mutations involve substitutions of the first-position glycine with cysteine or serine (Gly-X-Y). The other permissible substitutions are alanine, arginine, aspartate, glutamine, and valine. Substitution in the second or third position with a large branched chain (e.g., valine) or charged amino acids (e.g., aspartate or glutamate) is associated with more severe disease. C-terminal substitutions are more severe than N-terminal mutations because the pro-α chains assemble from the C- to N-terminal direction.
Fibroblasts and osteoblasts in patients with Type I OI produce one-half the normal amount of collagen as the result of a mutation altering one allele. This quantitative (null allele) effect results from a mutation inducing a premature termination codon affecting transcription of either the pro-α(I) or pro-α 2 (I) chain messenger ribonucleic acid (mRNA) so that only one-half of the normal amount of collagen heterotrimer is secreted into the extracellular space. An intracellular degradative process “nonsense mediated decay” rapidly eliminates mutated RNAs leading to both marked reduction in steady-state levels of mRNA from the mutant allele and a quantitative decrease in type I procollagen production . This mechanism is operative in cases of mild, type I OI and may in part determine the ability of these patients to heal fractures with relatively little deformity.
As reported by Marini et al. in an analysis of 832 COL1 mutations, helical splice site mutations occurred in 20% of OI patients leading to exon skipping, intron inclusion, or the activation of cryptic splice sites . Splice site mutations involving α-(I) were rarely lethal but they often lead to frameshifts and the mild type I phenotype. In α-2 (I), lethal exon skipping events were located in the carboxyl half of the chain. C-propeptide mutations in COL1A1 can also lead to haploinsufficiency as the mutated pro-α chain is retained intracellularly .
A second mechanism affecting collagen synthesis is the “qualitative defect” which involves structurally abnormal pro-α (I) chains that are assembled into heterotrimers which although defective, are secreted and incorporated into ECM . A third possible type of mutation, one affecting the regulatory portions (promoter, enhancer regions) of the COL1A1 or COL1A2 genes, has not been reported.
48.4.2
Type II osteogenesis imperfecta (perinatal lethal osteogenesis imperfecta)
The estimated incidence of lethal type II (Sillence) OI is approximately 10% of the total OI population: the true incidence is not known.
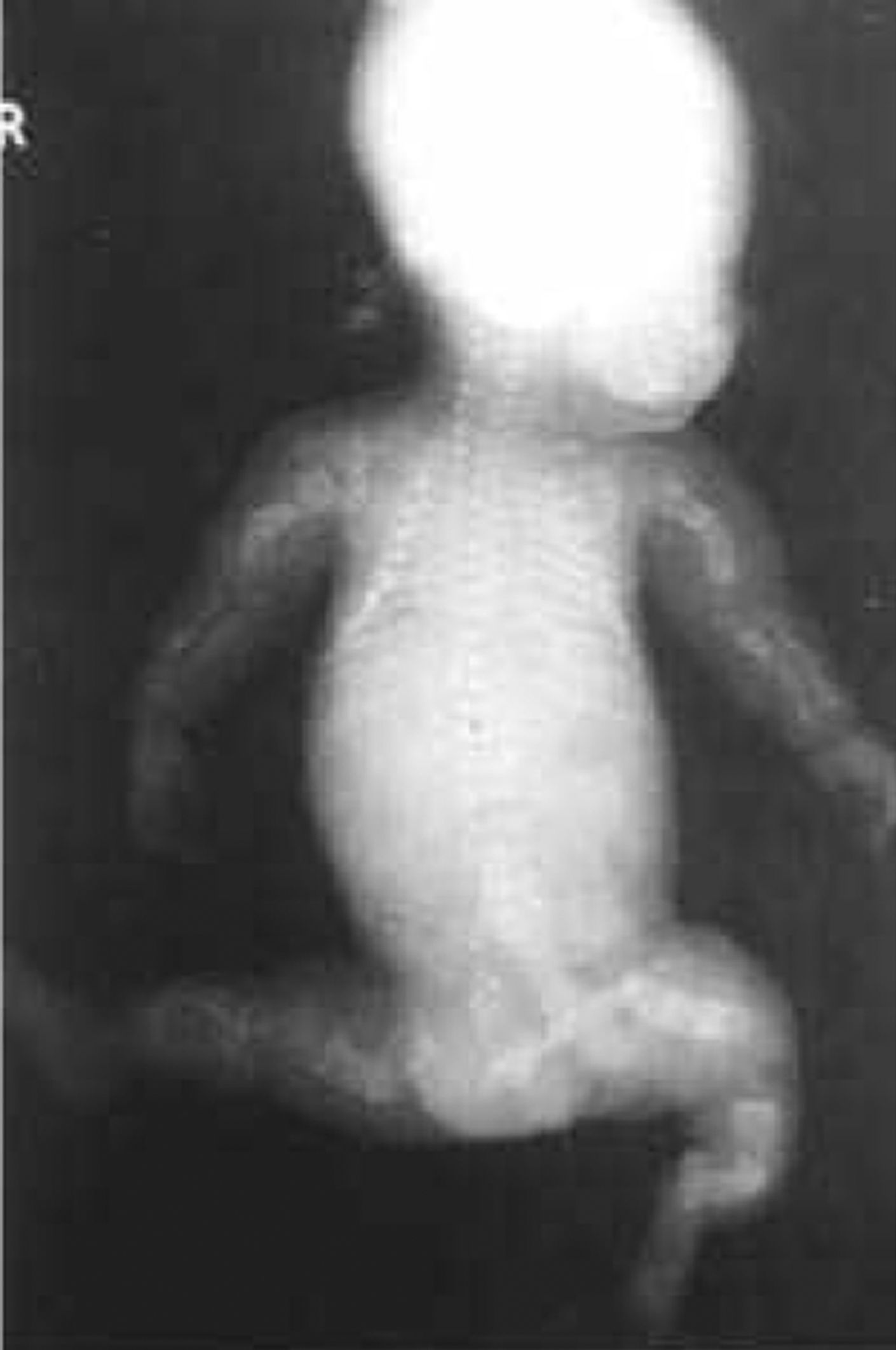
Type II OI has been classified into three groups by Sillence : based on modeling of the femurs and ribs. At birth, Group A have short, broad, “crumpled” long bones, angulation of tibiae and continuously “beaded ribs” due to callus on healing fractures ( Fig. 48.6 ). In group B, long bones were similar to group A but ribs were normal or had incomplete beading. In group C, long bones appeared long, thin, inadequately modeled (tubular) or rectangular with multiple fractures and thin beaded ribs. As discussed later, this phenotype overlaps that of recently described type VIII OI.
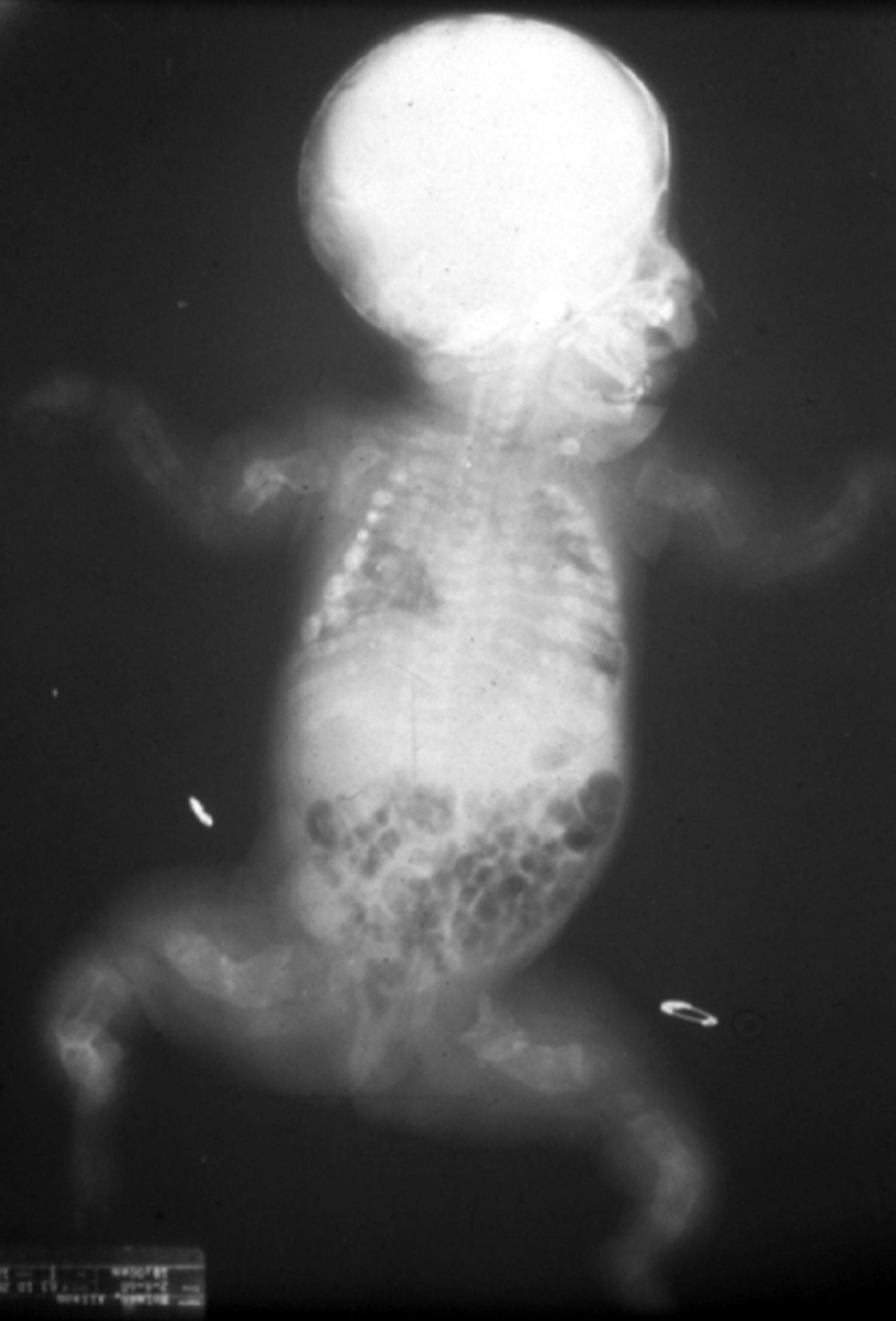
Many cases of type II OI are diagnosed early in pregnancy by three-dimensional ultrasound . Characteristic findings are deep blue sclera, shortening and bowing of long bone, fractures of long bone or rib with callus formation (beading), calvarial Wormian bones, nuchal translucency, frontal bossing, platyspondyly, and decreased bone mineralization . These infants are small for gestational age. Organ involvement is widespread, manifested as severe skeletal deformity with diffuse fracture involvement of the extremities, ribs, calvarium, and spine. Apgar scores are depressed at birth. The major life-threatening complication is pulmonary insufficiency due either to mechanical factors secondary to multiple rib fractures or to primary pulmonary insufficiency caused by pulmonary hypoplasia. Secondary complications include traumatic brain hemorrhage, spinal cord injury, and avulsion of body parts during delivery.
The differential diagnosis includes infantile hypophosphatasia, thanatophoric dwarfism, asphyxiating thoracic dystrophy, and achondroplasia. In hypophosphatasia, there is widespread failure to ossify due to low alkaline phosphatase (ALP) activity . In achondroplasia, the bones are short and tubular: mutations are found in fibroblast growth factor receptor-3 . Asphyxiating thoracic dystrophy is associated with a narrow or bell-shaped thorax and may mimic the pulmonary insufficiency of type II OI .
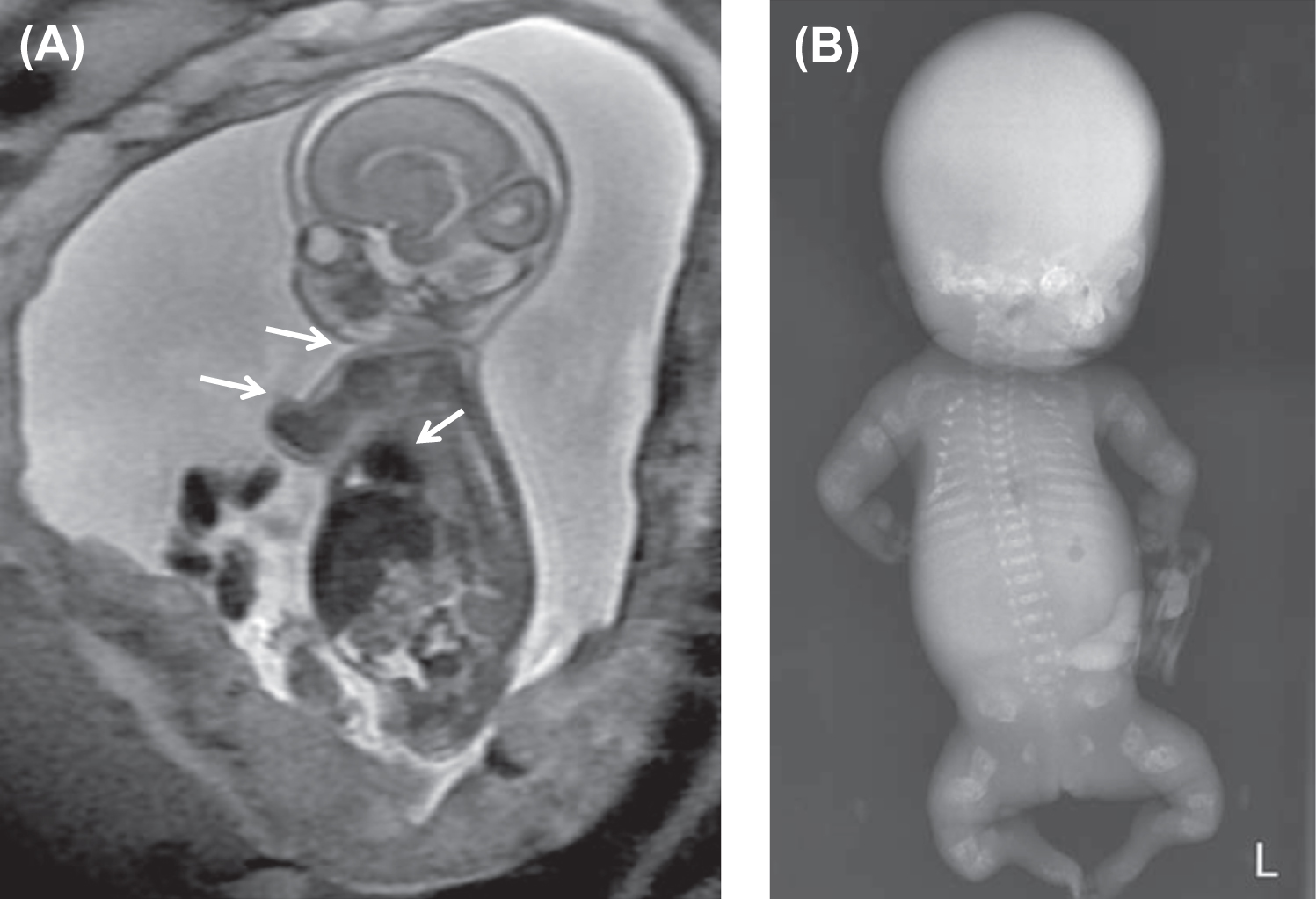
Fig. 48.7 illustrates distinctive magnetic resonance images of a type II fetus in utero and the postnatal skeletal radiograph . In OI type II the cranial vault is paper thin and may show considerable molding. Wormian bones, multiple small islands of underossified cranium, are visible in the occipital and parietal regions . The extremities appear foreshortened (rhizomelia) due to fractures and appear either broad and crumpled or tubular with a bulbous metaphysis containing whorls of calcified cartilaginous tissue (popcorn calcification). Clavicles and ribs contain multiple fractures. The ribs are typically narrow and sometimes exhibit a beaded appearance due to the presence of healing intrauterine fractures. The spine shows platyspondyly. Infants with type II OI either die at birth or survive for periods of days to weeks. Occasionally, infants will survive for several months depending on the available nutritional and ventilatory support. However, the integrity and maturity of the thoracopulmonary system usually determines the outcome, with infection always a risk.
The histology of bone in type II OI demonstrates markedly defective cortical and trabecular bone formation (woven bone). The process of endochondral bone formation at the epiphysis is disorganized, leading to persistent islands of cartilage and undermineralized bone. Membranous bone formation is similarly deficient, resulting in marked thinning of the calvarium. In two sporadic cases of OI type IIC termed “dense bone variant,” histological examination showed broad, interconnected cartilaginous trabeculae with thin osseous seams in the metaphyses. These cases showed deformed slender long bones with dense metaphyseal margins and normal vertebral bodies which contrasted with the crumpled, thick long bones and multiple vertebral compression fractures in OI IIA . Pace et al. have reported as lethal OI, a case in which the ends of long bones were not compressed but had regions of increased bone density. Histological studies identified decreased osteoclasts, abnormally thickened bony trabeculae with retained cartilage in long bones, and diminished marrow spaces . In contrast with these lethal cases, Lindahl et al. have reported C-propeptide cleavage site mutations in two patients associated with a mild OI phenotype where dual x-ray absorptiometry (DXA) measurements demonstrated high rather than low bone mineral density (BMD) .
48.4.2.1
COL1A1 and COL1A2 mutations associated with type II osteogenesis imperfecta
Advances in the definition of mutations indicate that the perinatal lethal OI phenotype (type II Sillence) occurs under two circumstances: first as a result of dominant mutations involving COL1A1 and COLIA2 and second as a result of recessive mutations involving the CRTAP gene that encodes cartilage associated protein located on chromosome 3, the LEPRE1 gene which encodes leucine proline-enriched proteoglycan (leprecan) 1 located on chromosome 1 and PPIB (cyclophilin B, CYPB) located on chromosome 15 . COL1A1 and COL1A2 mutations may occur (1) de novo where the parents are uninvolved, or (2) as a consequence parental mosaicism, and (3) due to recessive disease with carrier parents.
Mutations associated with the lethal form of OI frequently involve missense or nonsense single base substitutions, deletions, or insertions of various sizes affecting the C-terminal domains of the helical region or the C-propeptide of either the COL1A1 or the COL1A2 genes. However, as in phenotypes III and IV, splicing mutations, deletions, and insertions of central or N-terminal domains have also been described . It is of interest that similar mutations have also been associated with milder type I disease (see Collagen Mutation Database ). Unlike the intracellular decay associated with a null allele, assembly of the mutated pro-α chain with normal chains occurs, and the structurally abnormal heterotrimeric product is secreted into the ECM.
The occurrence of mutations in OI involving the helical domains in pro-α 1 (I) and pro-α 2 (I) were reported in 832 cases by the OI Consortium for Mutations . A total of 682 involved substitutions for glycine residues in the triple-helical domain and 150 altered splice sites. One-third of the glycine substitutions in pro-α 1 (I) was lethal, especially when the substituting amino acids were charged or had a branched side chain. Two regions associated with lethal OI (helix positions 691–823 and 910–964) aligned with major ligand binding regions (MLBRs), suggesting crucial interactions of collagen monomers or fibrils with integrins, matrix metalloproteinases (MMPs), fibronectin, and cartilage oligomeric matrix protein (COMP). By contrast, 20% of the mutations in COL1A2 were lethal. Lethal substitutions were located in eight regularly spaced clusters along the chain. As in COL1A1m the lethal regions aligned with proteoglycan binding sites along the fibril. Bodian et al. sequenced the coding and exon-flanking regions of COL1A1 and COL1A2 in a cohort of 63 subjects with OI type II ; 61 heterozygous mutations in type I collagen were identified. Of 34 COL1A1 mutations, 26 involved substitution for glycine within the Gly-X-Y triplet. Four mutations altered splice sites resulting in exon skipping: one resulted from a deletion spanning coding and intronic sequence, one was a nine-exon deletion from genomic DNA, one involved duplication of nine nucleotides, and one altered a single residue in the C-terminal propeptide. Twenty-two mutations were identified in COL1A2 resulting in glycine substitutions, deletions, and splice site mutations causing exon skipping .
Mutations in the type II OI “thick bone variants” have involved the C-terminal propeptide domain. Pace et al. reported a COL1A1 4321G→T transversion in exon 52 that changed a conserved aspartic acid to tyrosine (D1441Y) . In two OI IIC cases reported by Takagi et al., one sibling had a novel heterozygous mutation in the C-propeptide region of COL1A1, while no mutation in that region was identified in the second patient . In contrast to these type II patients, both COL1A1 and COL1A2 C-propeptide cleavage site mutations were associated with high BMD (DXA) measurements in two subjects reported by Lindahl et al. . These cases illustrate the lack of genotype/phenotype relationship observed in OI.
48.4.3
Type III osteogenesis imperfecta (severe, progressive osteogenesis imperfecta)
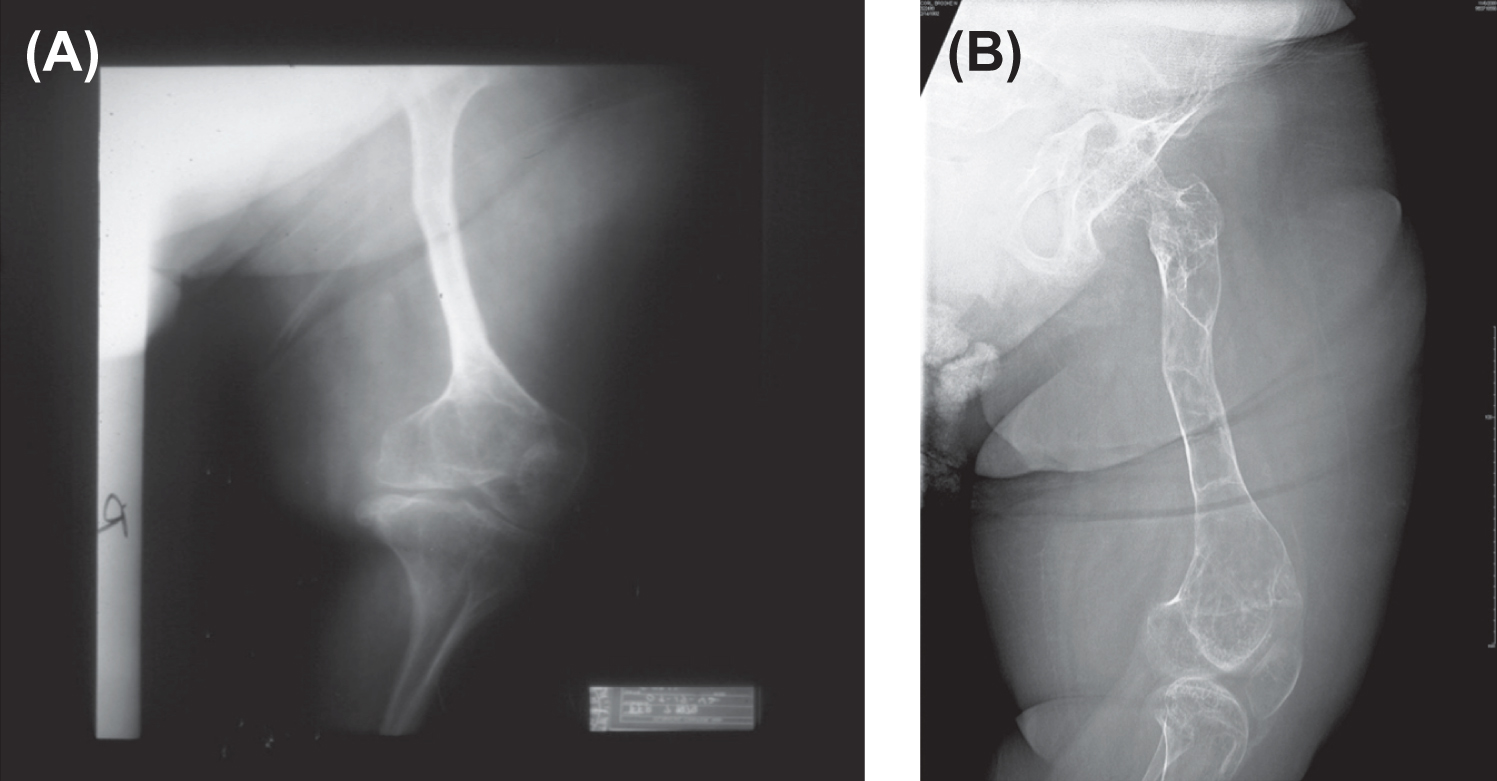
Infants born with severe OI present a characteristic appearance due to the presence of multiple fractures and deformities of the limbs. Birth weight and length are initially within normal, although retarded growth appears within the first year of life. Fractures at birth may involve the cranium, ribs, clavicles, and long bones. The cranium, although normal in circumference, appears relatively large. With growth, an occipital overhang or “helmutshadel” deformity of the calvarium may develop. Molding of the cranium alters facial proportions so that a “sunset” appearance to the eyes may occur. Type III malocclusion of the mandible causes mild prognathism. Sclerae are usually blue at birth but this decreases with age so that white sclerae are more typical of adults with type III OI. Moderate thoracic deformity with a pectus carinatum may be present but rib fractures are uncommon. Scoliosis may be mild initially but, with growth, approaches moderate-to-severe proportions ( Fig. 48.8A and B ). Multiple vertebral fractures may be present at an early age. Vertebral fractures and ligamentous laxity contribute to the progression of scoliosis.
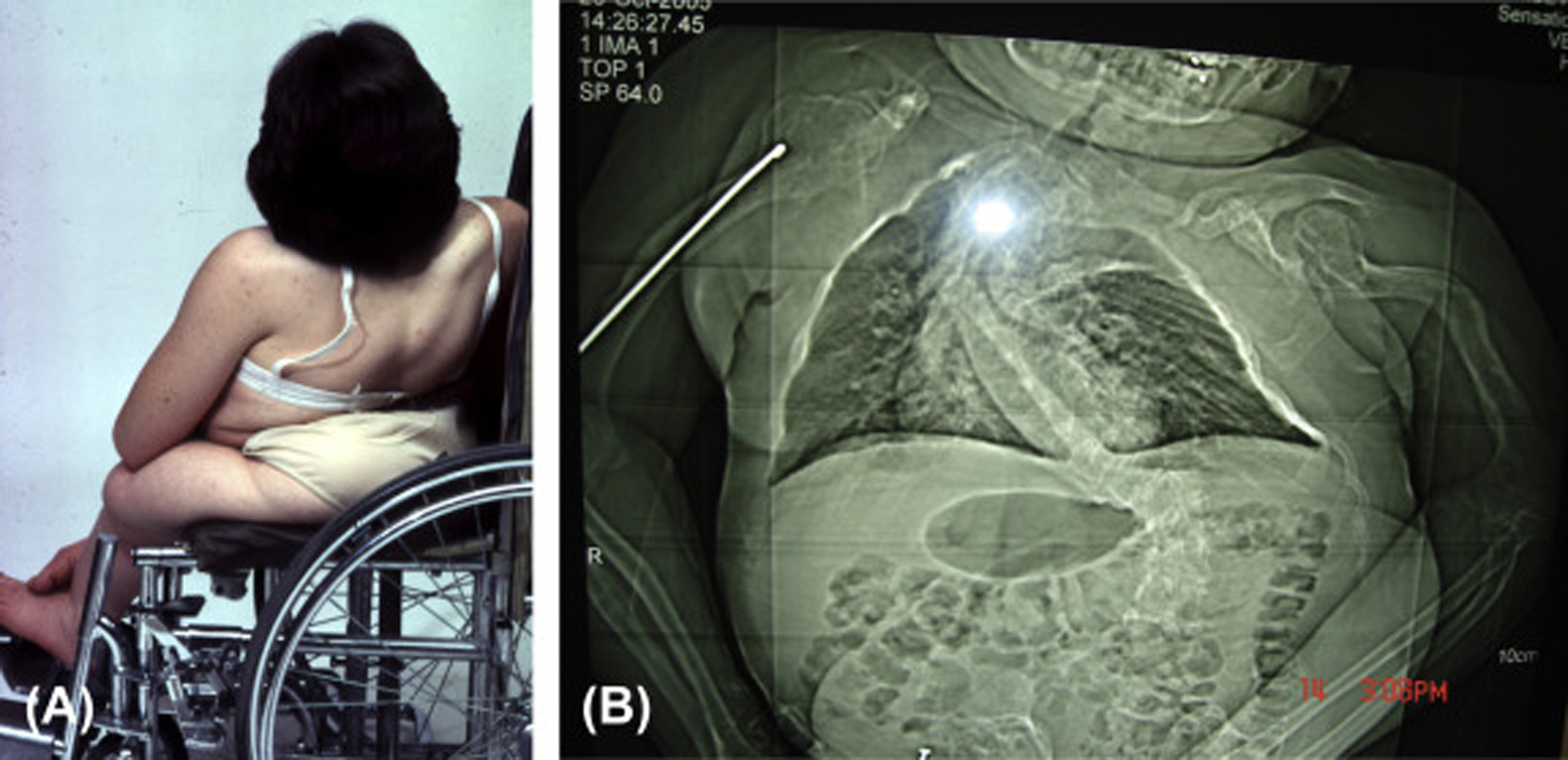
The limbs are deformed by the pull of muscles and ligaments on the undermineralized bone. Individuals with type III disease have a characteristic high-pitched voice. There is a profound failure of somatic growth, many patients reaching only 3 or 4 ft. (0.91–1.22 m) in height. Deformities of the upper and lower extremities are present from birth and aggravated following recurrent fractures. Because of skeletal deformities and severe osteopenia with a propensity to fracture, type III individuals tend to be wheelchair dependent. Complications in the adult include the ever-present risk of traumatic fracture, a syndrome of chronic headaches (occipital cough headache) related to basilar invagination, hearing loss, and progressive pulmonary insufficiency. A critical problem may occur if the medullary respiratory center or the cervical spinal cord is compromised by deformity related to basilar invagination. Following puberty, as is common with other OI types, the incidence of fractures declines markedly.
The radiologic appearance may be of either the “broad bone” or “narrow bone” type, both representing a severe defect in skeletal modeling ( Fig. 48.9 ). The epiphyses are poorly defined in these children, perhaps accounting for the limited skeletal growth. The epiphyses may contain irregular areas of poorly mineralized whorls of connective tissue (popcorn calcification).
48.4.3.1
Mutations in type III osteogenesis imperfecta
As in types I and II disease, a spectrum of mutations affecting various domains in the COL1A1 and CO1A2 genes have been reported. These are listed in the collagen mutation database ( http://www.le.ac.ul/genetics/collagen ).
48.5
Type IV osteogenesis imperfecta (OMIM 166,220)
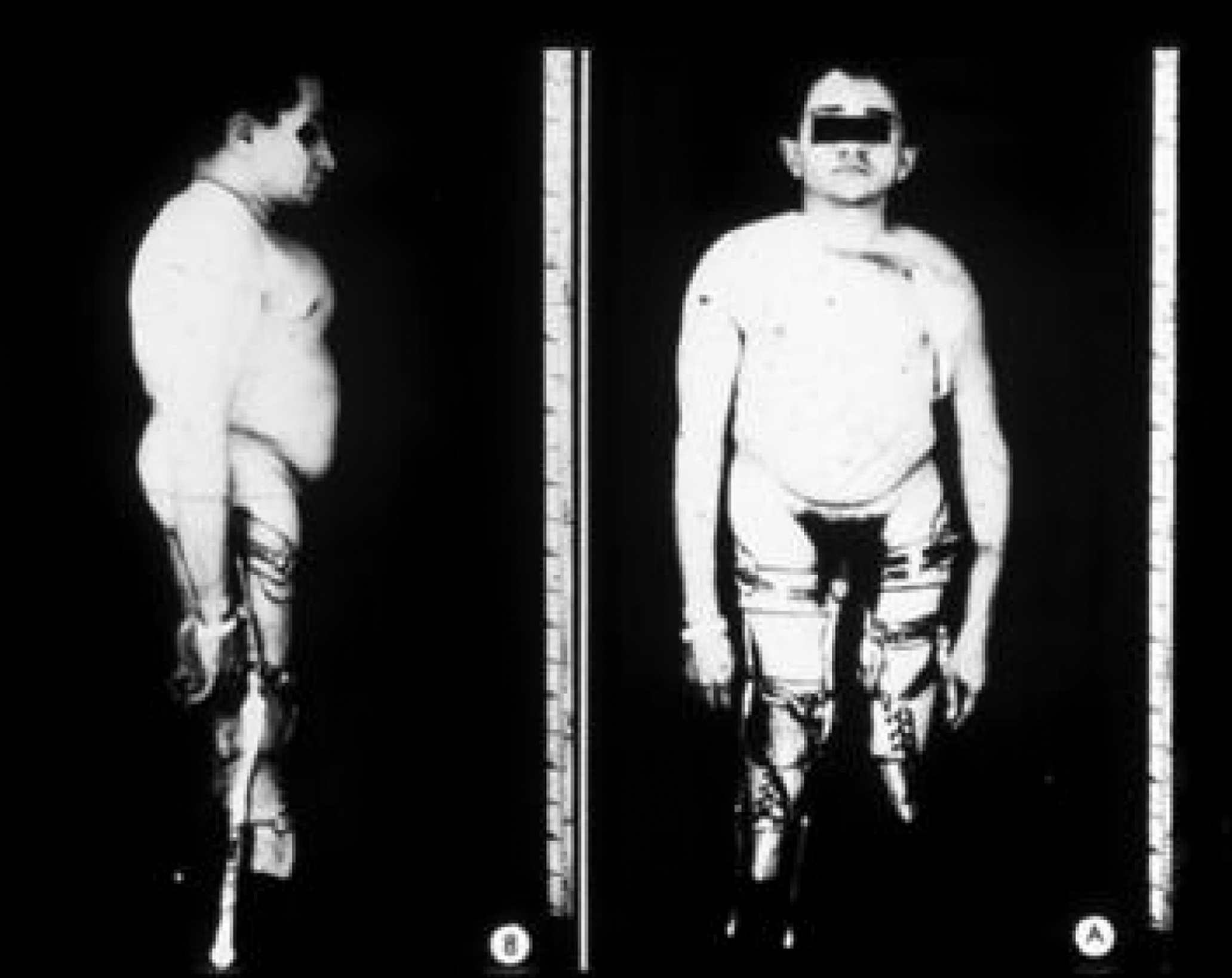
This phenotype, which includes approximately 15% of the OI population, is recognized as clinically heterogeneous. It was initially categorized by Sillence as moderately severe OI, characteristically having blue sclerae at a young age that faded to a white hue as adults . However, individuals with this phenotype may retain blue sclerae as adults. This phenotype is inherited as an autosomal-dominant trait. Both the mild and severe extremes of this phenotype may be confused with type I or III OI. Clinically, these individuals have short stature and a tendency to cranial molding, and DI may affect approximately 25% of cases. Molding of the calvarium persists into adulthood. Basilar impression and neurologic symptoms are reported to occur in 71% of type IVB OI patients . In type IV, both vertebral and appendicular bone are more osteoporotic and dysplastic ( Fig. 48.10 ). Scoliosis may be prominent. Pelvic deformity is common in these individuals. Joint laxity may disrupt the architecture of the ankle joint with a tendency to inversion, and dislocation of the knees may occur. Growth in height is intermediate between those with types I and III OI. In type IV OI, there is more extensive skeletal deformity than in type I disease and the osteopenia of underlying bone is more severe. As a consequence, many individuals rely on either a cane or crutches for ambulation.
48.5.1
Mutations in type IV osteogenesis imperfecta
Wenstrup et al. initially described mutations involving COL1A2 in type IV OI . However, both pro-α 1 (I) and pro-α 2 (I) chains may be mutated . In general, these mutations occur near the central region of the helical chain consistent with a gradient effect on phenotype. The initial report of a mutation in type IV OI involved the pro-α 2 (I) chain, subsequently defined as an exon 12 skip secondary to a G→T substitution affecting the consensus donor splice site . Intron mutations leading to an exon skip in type IV disease have been reported to involve pro-α 1 (I) exon 8 and pro-α 2 (I) exons 12 and 21 . The exon 21 skip was in a boy with short stature, osteoporosis, and dentinogenesis but no fractures . A pro-α 1 (I) gly352→ser mutation affecting the helical region was also reported . Interestingly, both pro-α 2 (I) gly646→cys and gly661→ser mutations have been reported in type IV disease . Clinical variability in a family with OI IV disease was explained as due to a COL1A2 mutation leading to alternative splicing of exon 26 . Novel COL1A2 mutations in type IV OI have been recently reported by Lin et al. .
48.5.2
Type V osteogenesis imperfecta
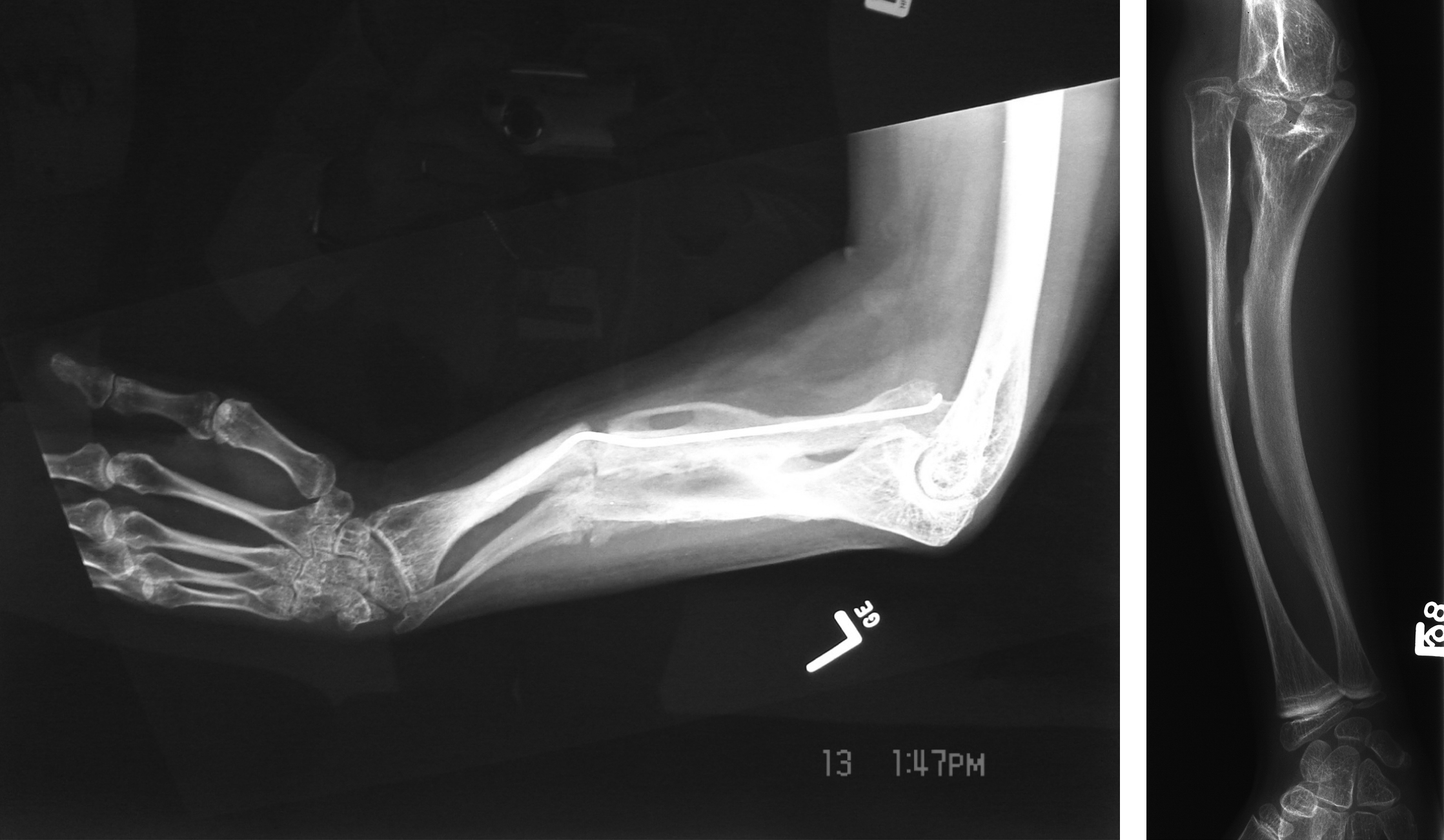
First reported by Glorieux et al. in 2000, OI type V is a nonlethal autosomal-dominant form of OI . Type V accounts for 4%–5% of the OI population. As with other nonlethal forms of the disorder, such as Sillence types I, III, and IV, OI type V has been characterized by moderate bone fragility, scoliosis, long-bone deformities, and diminished stature. However, type V demonstrates highly variable expressivity even within an individual family. It is distinguished from other OI types by the frequent occurrence of hyperplastic callus (HPC) following fracture or surgery (vide infra, HPC), the presence of bilateral radial-head dislocation, and ossification of the interosseous membrane in the forearm and lower extremity. However, type V patients do not express blue sclerae or DI.
Fig. 48.11A and B shows the characteristic interosseous calcification and radial head dislocation in type V OI. Calcification of the interosseous membrane can occur at an early age. Dislocation of the radial head is not apparent in infants but occurs during the first 3–5 years of life. X-ray findings include the presence of dense metaphyseal bands in long bones .
The histology of type V has been reported as showing an irregular mesh-like lamellar structure. Cortical and trabecular bone volumes are decreased on bone biopsy and dynamic parameters of bone formation are decreased . The type I collagen mutation involved in type V OI has recently been reported : this involves a heterozygous (c. −14C>T) mutation in the 5′-untranslated region of IFITM5 (the gene encoding interferon-induced transmembrane protein 5). Although this protein is expressed early in osteoblast development, the relation of this mutation to the phenotype is not defined at this time. The response to treatment with intravenous pamidronate appears similar in type V patients to that seen in OI types I–IV .
48.6
Recessive forms of osteogenesis imperfecta
48.6.1
Osteogenesis imperfecta type VI (OMIM 613,982)
OI type VI, initially reported by Glorieux et al., is a moderate-to-severe OI phenotype with marked skeletal deformity . Sclerae are white or faintly blue and DI was not observed. The diagnosis of OI type VI is largely based on bone biopsy histopathology which shows thin cortices, excessive osteoid deposition, and a prolonged mineralization lag time with a decrease in mineral apposition rate . In the initially reported series, there was a greater fracture risk including vertebral fractures compared to type IV OI. Also, signs of rickets were absent both clinically and radiologically. Serum ALP levels were elevated consistent with the mineralization defect. Treatment of this group with intravenous pamidronate did not correct the mineralization defect. Fracture incidence was decreased but not to the extent seen with other OI groups . DNA analysis indicates that OI type VI is the result of mutations affecting the collagen chaperone SERPINF1, the gene coding for pigment epithelium derived factor . However, the relationship between this gene product and the mineralization defect remains unexplained.
48.6.2
Osteogenesis imperfecta type VII (OMIM 610,682)
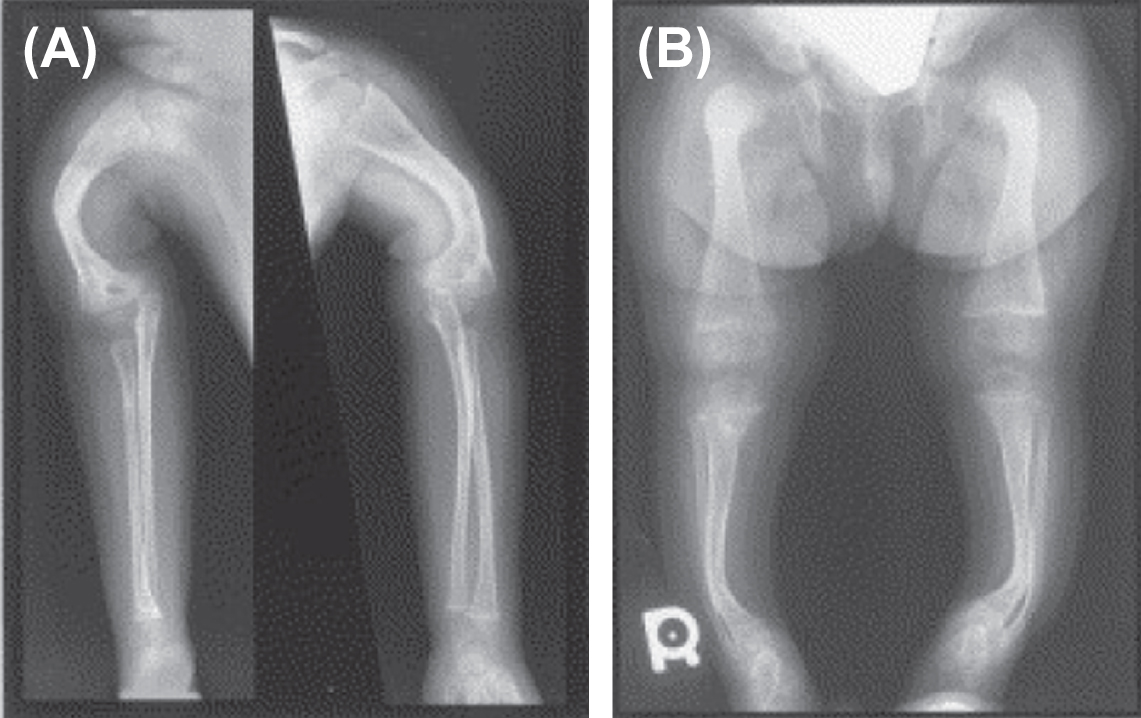
In 2002 Ward et al. described an autosomal-recessive, moderately severe OI type found in the First Nations Community in northern Quebec, Canada . Patients exhibited moderate-to-severe skeletal disease with deformities, blue sclerae, coxa vara, and osteopenia. Unlike the Sillence phenotypes I–IV, these patients had rhizomelia ( Fig. 48.12A and B ). Iliac crest bone biopsy revealed findings similar to OI type I which appears at variance with the clinical severity of the phenotype.
Heterogeneous patterns of mineralization have been observed in bone biopsies from type VII patients and in animal models. Quantitative backscattered electron imaging (qBEI) was used to assess bone mineralization density distribution (BMDD) in femurs from Crtap −/− mice and transiliac bone biopsies from four children with mutations permitting residual CRTAP expression . In Crtap −/− animals and OI type VII patients, bone matrix analyses revealed a significant increase in mean mineral concentration (CaMean) and most frequent mineral concentration (CaPeak) compared to wild-type littermates and control children, respectively. The relationship of the heterogeneous bone mineralization pattern to fracture risk remains to be defined.
Morello et al. have defined mutations involving CRTAP (cartilage-related protein) in patients with OI type VII . CRTAP forms a complex with P3H1 (LEPRE1) and cyclophilin B (CYPB) prolyl 3-hydroxylate at position 986 in the pro-al1(I) polypeptide chain. When mutated, prolyl underhydroxylation leads to slowing of posttranslational processing due to excessive lysyl hydroxylation, excessive glycosylation, and perhaps interference with assembly of the triple-helical domain of type I collagen. Using cultured fibroblasts expressing CRTAP deficiency, Valli et al. identified a severe deficiency (10%–15% of control) of collagen deposited in ECM, with disorganization of the minimal fibrillar network . Multiple CRTAP mutations have now been reported . As noted in this chapter, mutations in the CRTAP complex are associated with phenotypes that are moderately severe or lethal. The basis for these genotype/phenotype relationships remains to be defined.
48.6.3
Osteogenesis imperfecta type VIII (OMIM 610,915)
The phenotype of OI type VIII overlaps that of Sillence types II and III OI in being either severe or lethal ( Fig. 48.13 ) . These patients have severe osteoporosis with fractures at birth, rhizomelia, and thin calvaria with large fontanels. Similar to other type II and severe type III patients, these infants had white sclerae, a round face, and a short barrel-shaped chest. Long-bone radiographs of surviving probands showed undertubulated, narrow bones with bulbous metaphyses. Their hands appeared relatively long compared to their forearms, with long phalanges and short metacarpals. Their bone density was lower than almost all individuals with severe OI. This phenotype is caused by mutations in the LEPRE1 gene which encodes P3H1 responsible for 3-hydroxylation of proline-986 . It had been recognized that recessively inherited type III OI occurs in South African families . Cabral et al. studied five west African patients whose phenotype overlapped types II/III (lethal/severe) OI. This group reported mutations in LEPRE1 (P3H1) that led to minimal 3-hydroxylation of α 1 (I) proline-986. There was associated collagen overmodification with excess lysyl hydroxylation and glycosylation resulting in delayed collagen secretion .
48.6.4
Mutations in chaperone proteins that are expressed as an osteogenesis imperfecta phenotype
Chaperone proteins modify the procollagen peptides during transit in the endoplasmic reticulum. These include SERPINF1, which encodes pigment epithelium-derived factor (PEDF), and SERPINH1, CRTAP, P3H1, and PPIB, which also function as chaperones. Chaperone proteins facilitate protein folding and catalyze peptide and disulfide isomerization required for pro-α chain assembly . Defective chaperone function leads to the accumulation of insoluble partially folded polypeptide chains in the endoplasmic reticulum and initiates the endoplasmic reticulum stress response . In response to unfolded collagen polypeptides, the cell downregulates protein synthesis. Aggregated misfolded chains are targeted for lysosomal degradation . If severe, this unfolded protein response can promote apoptosis.
48.6.4.1
SERPINH1 (FKBP10)
Mutations in this gene which encodes the rough endoplasmic reticulum chaperone protein FKBP65 were initially reported by Alanay et al. in Turkish families and a Mexican family with moderately severe OI . Venturi et al. report the case of a patient with an initially mild and then progressively severe form of OI due to a novel homozygous splicing mutation in FKBP10 (intron 8 c. 1399+1G>A), which resulted in aberrant mRNA processing and consequent lack of FKBP65 chaperone .
Mutations in FKBP10 clinically overlap with the Bruck syndrome phenotype of joint contractures associated with bone fragility .
48.6.4.2
SERPINF1
Mutations in the chaperone SERPINF1 are associated with OI type VI which is characterized by deficient mineralization and hyperosteoidosis. The SERPINF1 gene encodes the PEDF, which belongs to the serpin family of peptidase inhibitors .
PEDF has multiple actions: PEDF expression is expressed in osteoblasts and to a lesser degree in osteoclasts, which is possibly related to the mineralization defect in OI type VI . PEDF is capable of inducing differentiation of precursor cells into mature osteoblasts. Additional actions of PEDF include improved neuronal survival and differentiation and inhibition of angiogenesis. PEDF is thought to play a central role in the development of the neural retina. PEDF was recently identified as an adipokine whose concentrations are elevated in type II diabetes and in the metabolic syndrome .
48.6.4.3
Bruck syndrome (BS-1: OMIM 259,450; BS-2: 609,220)
Bruck syndrome is a rare, recessively inherited disorder which combines skeletal changes resembling OI with congenital contractures of the large joints (arthrogryposis) . Contractures are the result of pterygia affecting multiple joints. Rib and long-bone fractures, vertebral fractures, and calvarial Wormian bones occur in Bruck syndrome patients. Affected individuals have white sclerae, normal dental quality, and normal hearing. Bruck syndrome-1 is associated with mutations affecting the endoplasmic reticulum chaperone protein FBK10 . Bruck syndrome-2 is associated with mutations affecting PLOD2 . PLOD2 mutations affect lysyl oxidase-2 hydroxylation in the telopeptide domain and result in altered folding of the procollagen molecules . Mutations in FBK10 are also associated with a moderately severe OI phenotype that does not include joint contractures .
48.6.5
Mutations in noncollagen-related proteins associated with the osteogenesis imperfecta phenotype
48.6.5.1
SP-7 (Osterix/OSX)
An Egyptian child with recessive OI presented with recurrent fractures, mild bone deformities, delayed tooth eruption, normal hearing, and white sclera. Homozygosity mapping and a candidate gene analysis revealed a homozygous single base pair (bp) deletion (c. 1052delA) in SP7/Osterix .
48.6.5.2
Bone morphogenetic protein-1
BMP1/Tolloid (TLD)-like metalloproteinases are essential to the formation of the ECM. These proteinases regulate the processing of different precursor proteins into functional enzymes, structural proteins, and proteins involved in initiating mineralization of the ECM. An Egyptian family with consanguinity had two children who were diagnosed with severe autosomal-recessive OI and a large umbilical hernia. Homozygosity mapping in this family showed lack of linkage to previously known OI-recessive genes but revealed a homozygous region on chromosome 8p in the two affected sibs, which comprised the procollagen I C-terminal propeptide endo-peptidase gene BMP-1. Mutation analysis identified a Phe249>Leu homozygous missense c mutation in both patients, within the BMP-1 protease domain .
48.7
Genotype expression in osteogenesis imperfecta
48.7.1
Inheritance patterns and osteogenesis imperfecta
There are no good data relating the incidence of sporadic versus familial occurrence of OI. Estimates for the prevalence of sporadic disease vary from 19% to 25% . As indicated later, the occurrence of mosaicism, where the parents have a normal phenotype, has made this estimate even more uncertain. For families in which dominant inheritance is based on a type I collagen mutation (types I–IV) the risk in successive pregnancies is 50%. For many years, approximately 25% of cases were thought to be recessively inherited. Data indicate that only 3%–5% of OI cases can be attributed to recessive inheritance, that is, the subsequent risk to parents of a child with sporadic severe or lethal disease (type VII or VIII) . In the case of gonadal or somatic mosaicism with dominant transmission, the risk on successive pregnancy would be 50%. Pyott et al. have surveyed the recurrence of lethal OI in sibships, that is, the recurrence rate of lethal OI after the birth of an affected infant . The recurrence rate regardless of genetic background in the parents after the first affected pregnancy was 1.3%. The rate of parental mosaicism in families in which a dominant mutation was identified in a first affected child was 16%. In 37 families with two affected infants, 26 had dominant mutations, seven had recessive mutations, and no mutations were found in four. With regard to lethal phenotypes, mutation identification allowed separation of families into one of three risk groups: those with a risk below 0.1% in the absence of parental somatic mosaicism or recessive inheritance, those with a risk of up to 50% with parental mosaicism, and those with a 25% risk if both parents are carriers of a recessive mutation .
48.7.2
The relation of genotype to phenotype in osteogenesis imperfecta
The majority of type I collagen mutations are unique: only relatively few have appeared in more than one family. It has proven difficult to formulate a cohesive theory to explain the relationship between specific collagen gene mutations and the highly variable OI phenotypes. Certainly, the phenotype will in part depend on the type of mutation (single-base substitution, deletions, insertions, etc.), the substituting amino acid, and its location in the collagen molecule. Because the molecule assembles from the C- to the N-terminal direction, C-terminal mutations tend to be clinically more severe than N-terminal mutations. However, this rule is breached by several examples of mutations that have inconsistent effects on the resulting phenotype both within and among affected families. One explanation gleaned from the effects of different mutation loci on the patterns of thermal unfolding of type I collagen suggests that specific domains (cooperative melting domains) of the procollagen chains constitute regions specifically susceptible to altering chain assembly or stability . Similar mechanisms may explain the manner in which like mutations located at adjacent loci have dramatically different effects on the expressed phenotype. For example, deletion of exon 11 produces a phenotype like EDS without significant bone disease, while deletion of exon 12 produces type IV OI.
The Consortium for Mutations in the Helical Region of Type I Collagen surveyed 832 independent mutations, 682 resulted in substitutions for glycine residues in the triple-helical domain, and 150 altered splice sites . One-third of the glycine substitutions in α 1 (I) was lethal, especially when the substituting residues were charged or had a branched side chain. Two lethal regions (helix positions 691–823 and 910–964) aligned with MLBRs, suggesting significant interactions of collagen monomers or fibrils with integrins, MMPs, fibronectin, and COMP. Mutations in COL1A2 were predominantly nonlethal (80%). Lethal substitutions were located in eight regularly spaced clusters along the chain. In α 2 (I), lethal exon skipping events were located in the carboxyl half of the chain. These lethal regions also aligned with proteoglycan binding sites along the fibril.
Splice site mutations that lead to exon skipping comprise 20% of helical mutations identified in OI patients. In COL1A1 splice site mutations were associated with a milder phenotype .
Ben Amor et al. have evaluated genotype/phenotype relationships in patients with autosomal-dominant inheritance and type I collagen mutations . The following summarizes the results of this study:
- 1.
Glycine-to-serine substitutions were the most frequent type of mutation in the triple helix domains of the collagen type I α 1 and α 2 chains.
- 2.
Serine substitutions tended to a more severe phenotype in the α 1 than in the α 2 chain.
- 3.
The clinical severity of serine substitutions correlates with the position of the mutation in the α 2 chain but not in the α 1 chain.
- 4.
Substitutions by arginine, aspartate, glutamate, and valine beyond the first 200 amino acid residues of α 1 are generally lethal but have variable outcome in α 2 .
- 5.
Mutations affecting the first 120 amino acids of the collagen type I triple helix lead to blue sclerae but do not cause DI.
- 6.
Glycine substitutions in α 1 almost always are associated with the presence of Wormian bones, whereas about three-quarters of patients with glycine substitutions in α 2 and one-quarter of patients with haploinsufficiency mutations have Wormian bones.
- 7.
More than one-third of patients with glycine substitutions in the α 1 or α 2 chain but only 1 in 20 patients with haploinsufficiency mutations have cranial base abnormalities.
- 8.
Mutations at the C-terminal end of the α 2 chain are associated with limb anomalies and intracranial hemorrhage.
- 9.
No correlation was found between the mutated gene or mutation type and the hearing pattern in OI.
These studies, while informative, do not provide the predictive power that would be clinically applicable to the management of children or adults with OI.
48.8
The pathophysiology of osteogenesis imperfecta
48.8.1
Osteoblast metabolism in osteogenesis imperfecta
The osteoblast developmental cascade proceeds from bone marrow osteoprogenitor stem cells to preosteoblasts and terminally differentiated osteoblasts that do not further divide. Osteoblasts give rise to bone lining cells and osteocytes. Cultured human osteoblastic cells may be grown as explants from minced trabecular bone and subjected to metabolic study for as long as 35 days in tissue culture. These cells do divide and are at the preosteoblastic stage . An assessment of expressed osteoblastic markers by cultured cells from subjects with different OI types has revealed that the production of osteocalcin in response to 1,25(OH) 2 D 3 stimulation was similar in OI types I, III, and IV versus controls . However, both osteocalcin production and cAMP response to 1,25(OH) 2 D 3 were decreased in cultured type II OI osteoblasts as contrasted with fetal controls.
Several metabolic abnormalities related to ECM synthesis have been reported when cultured osteoblasts from OI subjects are compared to age-matched healthy people . Although the growth curve of cultured human osteoblasts is slower than that of normal fibroblasts, OI osteoblasts (but not OI fibroblasts) have slower rates of proliferation when compared to age-matched healthy people . The synthesis of type I collagen as measured by [ 3 H]proline incorporation is decreased in cultured OI osteoblasts as is the synthesis of the matrix associated proteins osteocalcin and osteonectin. The synthesis of the matrix proteoglycans decorin, biglycan, and the large chondroitin sulfate proteoglycan is also decreased . However, the synthesis of hyaluronan and bone sialoprotein by OI preosteoblasts appears to be increased, consistent with the increased amounts of bone sialoprotein and osteocalcin isolated from OI bone . Osteonectin content was also reduced in OI bone. It is assumed that these defects are secondary to the type I collagen mutation that in some manner alters the synthesis of other matrix proteins. However, the bone decorin levels were not altered in contrast to the observed decrease in decorin synthesis by OI osteoblasts in tissue culture. Collagen matrix deposition by cultured OI dermal fibroblasts from a spectrum of OI phenotypes was also decreased, indicating that diminished collagen secretion is not limited to subjects with the null allele phenotype .
Studies of collagen ECM deposition and turnover by fibroblasts from a case of lethal OI reveal the limitations on collagen secretion by these cells . OI fibroblasts with a gly667→arg mutation deposited one-quarter the type I collagen compared to controls. However, the reduction in collagen matrix deposition was not due to decreased total collagen synthesis; rather, the incorporation of the mutant collagen into matrix was less efficient. Such mutant collagen as incorporated into matrix appeared more subject to proteolytic breakdown, suggesting faulty copolymerization of the mutant with the normal procollagen.
Analysis of OI bone for collagen cross-link content has provided divergent results. Although decreased levels of lysine-based cross-links in skin have been reported , analysis of OI bone from subjects with types I, III, and IV OI for the mature collagen cross-links, hydroxylysylpyridinoline, and lysylpyridinoline, disclosed similar concentrations in OI and normal subjects .
An analysis of compact bone from 30 patients aged 2–9 years with various OI phenotypes demonstrated low collagen and low total protein content per milligram of DNA in OI compared to that in age-matched controls . This analysis also found increased content of type V but not type III collagen in OI compact bone. Type III collagen is not normally present in adult bone. However, increased types II and V collagen were found in collagen extracted from OI infants with lethal disease . In contrast to type I subjects where overhydroxylation of collagen was not found, posttranslation overmodification was found in bone collagen from subjects with types I and III OI.
Hydroxyapatite crystal size in bone specimens from children and adolescents reveals reduced c -axis crystallinity of apatite in types III and IV OI specimens and reduced crystal size during childhood only in type I OI subjects . It was postulated that reduced crystallinity was in some manner related to the defect in collagen synthesis because crystal size appeared reduced in more severely affected children. Reduced bone apatite crystal size had previously been demonstrated in two strains of cattle reported as having a phenotype that included blue sclerae, marked joint laxity, and osteoporosis, but in these animals, type I collagen synthesis was normal. The mechanism of altered crystal size in OI thus remains uncertain.
48.8.2
Histomorphometry of bone in osteogenesis imperfecta
The abnormalities in bone histology and histomorphometry generally parallel the severity of the OI phenotype. However, interpretation of bone histology is subject to qualification depending on the site of the biopsy, including proximity to an area of recent trauma, patient age, and the influence of medication that may alter bone turnover. Thus, to a varying extent, it is characterized by decreased trabecular volume and diminished cortical width, reflecting deficient matrix formation. There is mild increase in unmineralized osteoid. A striking finding in OI is the presence of increased numbers of osteocytes embedded in trabecular bone ( Fig. 48.9 ). This has been confirmed by direct counting of osteocytes in type I subjects (J. R. Shapiro, unpublished data). Increased numbers of osteocytes have also been reported in mild OI . Although plump osteoblasts are readily identified along trabecular margins, there is no increase in osteoclastic bone resorption. Electron microscopy of osteoblasts in OI has demonstrated dense material in the Golgi apparatus, glycogen deposits, and decreased ALP in the cell membrane .
The impact of a type I collagen mutation on endochondral bone formation can be seen in the growth plate in severe cases of OI (type II, severe type III). Cartilage columns appear to develop normally up to the point that endochondral bone formation occurs. In type II OI, there is a failure of normal lamellar bone formation. Rather than the bony trabeculae normally present at the time of birth, there are disorganized islands of cartilaginous core surrounded by islands of poorly mineralized woven bone.
Tetracycline-labeled iliac crest bone biopsies from 70 children, aged 1.5–13.5 years, were assessed by Rauch et al. . Bone core width and cancellous bone volume were decreased in all OI types. Trabecular number was decreased more than trabecular thickness. Production of secondary trabeculae by endochondral ossification was decreased. Bone remodeling balance was decreased in type I OI compared to controls but was very low in type III and IV OI. Matrix mineralization was normal.
Scanning electron microscopy (SEM) and transmission electron microscopy (TEM) and electron diffraction studies of OI bone have disclosed abnormalities in hydroxyapatite crystal size as well as in the morphology of type I collagen fibers and the organization of lamellar plates in bone. By SEM, type I OI bone did not differ significantly from normal in ultrastructure . In severe OI the lamellar structure of bone is disconnected and separated by open spaces in regions. Type II OI bone presented a spongy appearance. TEM of OI type III bone displayed a matrix of loose fibrous mineral that was undermineralized with abnormally oriented small crystals, poorly organized in relation to collagen fibrils. However, even in severe phenotypes, normally oriented lamellar bone structure may still be preserved.
Electron diffraction analysis of mineral crystals and direct measurement of crystal length from OI bone show that these are small in size and may be smaller in more severe phenotypes . The orientation of crystals in collagen fibers may also be abnormal. X-ray scattering studies of crystal orientation in bones from animals with the OI murine mutation have also revealed faulty orientation of crystals in collagen fibrils .
Immunohistochemical studies of bone from type II OI subjects have demonstrated nests of cartilage with type II collagen and increased amounts of type III collagen in bone matrix. The persistence of type III collagen mimicking a fetal pattern was shown in both type III and IV OI bone.
48.8.3
Animal models of osteogenesis imperfecta
Several animals of different species have been described as having the OI phenotype based on the presence of osteopenia with findings indicative of a connective tissue dysplasia. One example is the murine fragilitas ossium (fro/fro mouse) mutation developed as a result of treatment of male germ cells with tris(1-aziridinyl phosphine) sulfide . Collagen metabolism was normal in this model. A second model, termed bovine OI, was reported in the offspring of unrelated Friesian cattle in Australia and Holstein cattle in Texas . These animals had intrauterine or postnatal fractures, blue sclerae, and severe joint laxity. Although bone proteoglycan and sialoprotein content were depressed, type I collagen synthesis was normal .
The application of transgenic methodology has led to the development of several models of human OI based on mutations in the type I collagen molecule. In the MOV 13 heterozygous mouse model integration of a murine retrovirus in the first intron of COL1A1 blocks transcription simulating a null allele phenotype . A transgenic model containing a 45-bp pro-α 1 (I) deletion, first described in a lethal case of human OI, has proved viable . This minigene was designed to generate shortened pro-α 1 (I) chains to mimic the mutation, leading to intracellular protein degradation. In this model, 6% of the offspring died, 33% had fractures, and 61% had no fractures at birth, suggesting mosaicism .
A widely used animal model, oim/oim , is a viable, naturally occurring murine model that duplicates the phenotype of moderately severe human OI . This animal has a single base deletion in the C-terminal propeptide (exon 52) that prevents synthesis of pro-α 2 (I) and leads to the accumulation of pro-α 1 (I) 3 homotrimer in all tissues. Interestingly, it is not lethal despite the location of the mutation. Osteoporosis, fractures, joint laxity, and diminished somatic growth are features common to human OI. The mutation, a single base deletion, mimics a four-bp deletion in the same exon (exon 52) in a child with type III OI .
The Brtl IV murine model for OI is a heterozygous knock-in model carrying a Gly-349→Cys substitution in one ColA1(I) allele . Osteoblast function is decreased in this model. In addition, bone mass is decreased based on an imbalance between poorly functioning osteoblasts as well as increased RANK-expressing osteoclast precursors . It replicates moderately severe OI.
A new mouse model for OI termed Aga2 (abnormal gait 2) was isolated from the Munich N -ethyl- N -nitrosourea mutagenesis program. This model has reduced bone mass and multiple fractures. The gene was mapped to Chromosome 11 and a C-terminal frameshift mutation was identified in the Col1a1 gene as the cause of the disorder .
48.8.4
Bone mineral density in osteogenesis imperfecta
Normally, bone mass is determined by genetic, hormonal, and lifestyle factors. Most OI subjects have diminished bone density (osteoporosis) by X-ray, although both in children and in adults, bone mass may occasionally appear radiologically normal . DXA measurements in OI have demonstrated both decreased bone mass and the ability of mineral mass to increase to a limited extent with age in some subjects. Both trabecular bone density (vertebral, wrist, ribs) and cortical bone density (humerus and femur) are decreased.
High-resolution peripheral quantitative computed tomography (HR-pQCT) at the distal radius and distal tibia and DXA scans of total hip, femoral neck and the lumbar spine have been performed in 39 type I OI patients . Areal BMD was 8% lower at the hip and 13% lower at the spine compared to controls. pQCT scans indicated that volumetric BMD was 28% lower in the distal radius and 38% lower in the distal tibia.
In OI, bone mass will decrease during pregnancy and after menopause and after age 50 years in men. The fact that fracture rates increase after the menopause and with increasing age in men indicates the need for periodic monitoring of BMD to assess the rate of bone loss .
48.8.5
Bone turnover in osteogenesis imperfecta
The issue of bone turnover in OI is important as it relates to selection of treatment. For example, a bisphosphonate might be recommended where bone turnover is high and avoided in the presence of very low bone turnover. Ramser and Frost examined bone turnover in the rib of a woman with type I OI and determined that cortical bone turnover was increased three-fold, while that in the periosteal layer was diminished . This discrepancy between cortical and periosteal bone formation was unresolved but was proposed to contribute to diminished width of the ribs. Albright observed that the surface involved in new bone formation was increased as was resorption and the size of osteocytic lacunae . The presence of osteocytic resorption in OI bone has not been confirmed.
Bone turnover has been studied using double tetracycline labeling prior to biopsy. Ste-Marie analyzed iliac crest bone biopsies following tetracycline labeling and observed that in types I and IV OI, trabecular bone volume was decreased and the calcification rate was significantly reduced. Furthermore, reduced bone formation at the cellular level was evident in that the thickness of osteoid seams was low or normal in all subjects. In adults, there was no significant increase in the parameters of bone resorption or in the bone formation rates at the basic multicellular unit and the tissue levels. These results in adults may relate, as discussed later, to an impaired treatment response to bisphosphonates in adult patients with OI. Studies in children aged 6–15 years with mild OI demonstrated increased turnover rate with decreased osteoblastic activity . Glorieux et al. obtained iliac crest bone biopsies from 44 OI children aged 2–14 years with nonlethal disease (17 type I, 10 type III, 12 type IV) . Common to all OI types were decreases in cancellous bone volume, cortical width, and trabecular thickness. Cortical organization (lamellar matrix and Haversian systems) was impaired in relation to severity. Resorption activity and osteoclast number were not increased: there was a mild increase in eroded surfaces in type II OI. Mineral apposition rate was decreased for all OI types and significantly for types I and IV OI. Bone formation rate was decreased in type IV OI. Histomorphometric evidence of low bone turnover has also been observed in a cohort of type I OI adults . The observed differences in turnover rates between these recent results and earlier studies noted earlier may be due to improved technology but more likely result from consistent clinical classification not previously available.
48.8.6
Biochemical markers of bone turnover in osteogenesis imperfecta
Biomarkers of bone formation, serum osteocalcin, serum C-terminal procollagen type I propeptide (PICP), serum N-terminal PICP (PINP), and bone specific ALP have been measured in OI as have markers of bone resorption, urinary excretion of deoxypyridinoline cross-links, and the collagen N-telopeptide cross-link . These are potentially important because of histologic data suggesting that bone turnover was generally increased in OI children . Brenner et al. found PICP levels decreased in various forms of OI and more so in type I subjects . Osteocalcin levels were increased in patients during the first decade, but in only 1 of 18 older patients. In a subsequent report, elevation in deoxypyridinoline was reported, suggesting that increased resorption was a factor in the osteopenia of OI . However, increased resorption as a contributing factor to osteopenia in OI is not supported by histomorphometric analysis of bone in children or adults and has been disputed by Minisola et al. and Shapiro, who find markers of both formation and resorption commonly (but not uniformly) decreased in OI .
Bone biomarkers were assayed in 64 adult patients, mean age 36.2 years, by Garnero et al. Compared to controls, adults with OI had decreased levels of PINP (22.7%), increased osteocalcin (73%), and increased Col I helical peptide reflecting collagen breakdown (58%). Urinary a-CTx (C-terminal collagen cross-links) was increased +31%, whereas urinary and serum p-CTx were significantly decreased resulting in a 49% higher urinary a/p-CTx ratio. Patients with Col I gene mutations resulting in haploinsufficiency had lower PINP levels than patients with helical domain alterations and controls .
Bone specific ALP, osteocalcin, and PINP were measured in 24 type I OI patients (mean age 37±15 years) and compared to 25 patients with low bone mass due to other causes, and 38 controls . The median value for PINP was significantly lower in the OI group than in normal individuals. The median value for osteocalcin was significantly higher in the OI patients than in controls. Median values for bone ALP were significantly higher in both the OI and other low bone mass groups than in controls. The osteocalcin/PINP ratio was found to be a sensitive and specific test for type I OI in adults, but was less predictive for the diagnosis of other types of nondeforming OI (e.g., OI type IV). However, in contrast to the experience in adults, in groups of OI children, OI types I, III, and IV, aged 0.25–20 years, individual differences in bone biomarkers were not clinically useful. Significant differences in bone markers were found in the larger untreated group but not between subgroups with or without vertebral compressions. Pamidronate treatment caused a decrease in biomarkers during treatment for 1.0–12.5 years but to different relative amounts for each marker. Changes were not correlated to the improvement in BMD, mobility or pain . In summary, measuring bone biomarkers provides variable results in children and adults. However, a decrease in biomarkers is a consistent finding which serves as an index of the rate of bone turnover suppression during treatment with bisphosphonates.
48.9
Organ involvement in osteogenesis imperfecta
48.9.1
Ocular features of osteogenesis imperfecta
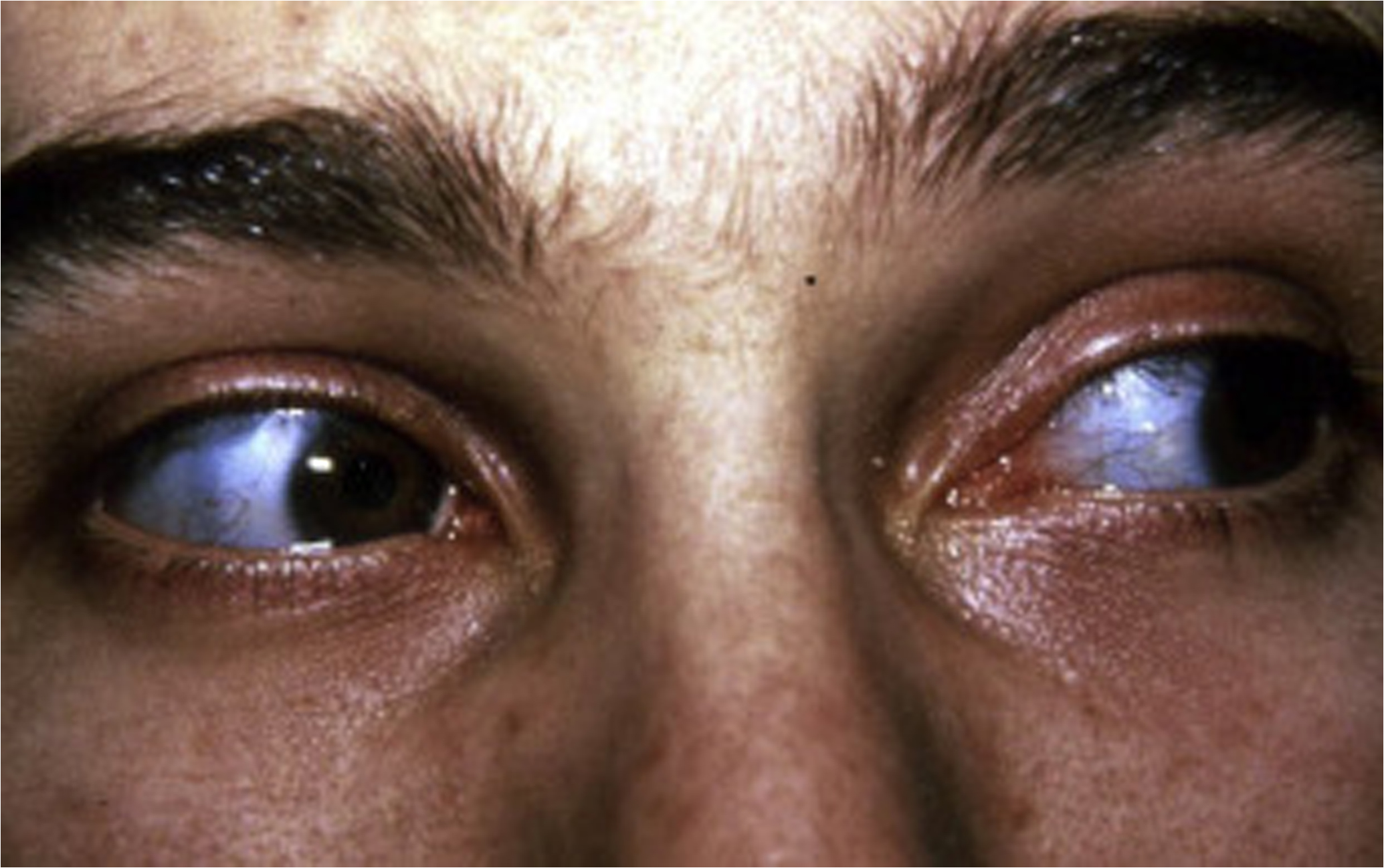
Scleral color may be a distinguishing feature of certain OI types; however, it is important to note that scleral hue may vary during a subject’s lifetime and that among connective tissue disorders, blue sclerae are not unique to OI . Blue sclerae have been described in subjects with the EDS, including unusual cases of human dermatosparaxis , Marfan syndrome (MFS) with contractures , and lethal hypophosphatasia . Blue sclerae occur uniformly in subjects with type I disease. Sclerae may be blue or white in individuals with type III disease and are frequently blue at a young age, fading to white in those with type IV disease.
Although the cause of blue scleral color remains unknown, it may be related to a matrix abnormality of the scleral coat since a positive relationship between the intensity of color and deformability of the globe has been demonstrated as well as decreased corneoscleral rigidity . In terms of physical characteristics the blue color is a product of reflectance rather than absorbance, again suggesting that it is dependent on altered matrix composition. Thin scleral coat as a cause of blue sclerae has not been a consistent finding, although Chan et al. have reported that in lethal OI, both corneal fiber diameter and the diameter of scleral collagen fibers were reduced by 25% and 50%, respectively . Arcus senilis (embryotoxon) is the second most frequent abnormality in OI following blue sclerae, being observed in 28% of affected individuals . It may occur as an opacity or arcus at the periphery of the cornea at a young age and appear as annulus senilis in an older individual. The cause of the lesion is unknown: it is not related to abnormal lipid metabolism. Specifically, ophthalmological evaluation of the OI patient should include measurement of central corneal thickness as related to the diagnosis of OI. Central corneal thickness is decreased and is negatively correlated with the blueness of the sclera in patients with OI .
Preliminary reports indicate that there is an increased incidence of glaucoma in OI. Isolated instances of several other ocular abnormalities have been reported in OI . These include myopia, rare instances of subluxed lens, keratoconus, and congenital Bowman’s layer agenesis .
48.9.2
Dentinogenesis imperfecta
Two dental lesions have been recognized in OI, DI, and multiple radiolucent bone cysts, which is a rare occurrence. Associated defects in the maxillofacial bone include condylar deformities with dislocation of the mandibular condyle, prognathic mandible (type III malocclusion), hypoplastic hemimandible, and depressed zygoma .
The most common oral manifestation of OI is DI. There are two types of DI. That common to OI is DI type II . Although DI occurs in approximately 15%–25% of each OI type, it is more frequent in type III OI and uncommon in type I OI. As a rule, DI tracks with bone disease so that individuals with DI in a family should be evaluated for bone disease. Affected teeth demonstrate a bulbous crown and increased coronal angle and may lack pulp space. Permanent teeth are less severely affected than deciduous teeth. Electron microscopy of dentin shows disorganization of dentinal tubules . It is this defect in dentin that interferes with the adherence of enamel to dentin and leads to chipping and erosion of the tooth.
Bone cysts of the jaw occur infrequently. A report describes a 23-year-old woman with OI and DI who developed multiple unilocular bilateral radiolucent cysts of the mandible 5 years after a condylar fracture .
48.9.3
Hearing loss in osteogenesis imperfecta
Diminished audioacuity is a frequent manifestation of OI having been recognized by testing in approximately 30% of all OI groups. Multiple functional lesions have been described. These include conductive defects and mixed or sensorineural lesions. Conductive loss is due to traumatic defects in the stapes crura or to fibrosis at the stapes footplate. The high incidence of mixed defects and sensorineural loss implies involvement of the cochlea or cochlear nerve . High-resolution CT and scintigraphy of the labyrinthine capsule has been performed in nine subjects with OI . A severe decrease in bone density in the pericochlear region was found in subjects with mixed hearing loss although normal density was found in other affected individuals. Tympano-cochlear scintigraphy suggested increased bone metabolism in this region. No relationship is reported between hearing deficits in OI and type I collagen mutations .
Audiological evaluation was conducted in 41 individuals at the Kennedy Krieger Institute . Forty-one patients with OI were included in the study. The patients were divided into two groups, one group less than 20 years of age ( n =21) and the other group aged 20 years and over ( n =20). Hearing loss of all etiologies was observed in 62% of ears. Sensorineural or mixed hearing loss was observed in 41% and conductive hearing loss in 21% of ears. The results indicated that the younger patients with OI were subject to a greater risk of middle ear dysfunction associated with otitis media than is typical for children of comparable age. Tympanometric abnormalities associated with ossicular dysfunction were more often found in the older age group of patients. Hearing loss of all types was more prevalent in the older group of patients (88%) than in the younger patient group (38%).
Swinnen et al. studied 182 OI patients, aged 3–89 years. Approximately 52.2% of all OI patients demonstrated hearing loss unilaterally (7.7%) or bilaterally (44.5%). Pure conductive, mixed, and pure sensorineural hearing losses were observed in 8.5%, 37.8%, and 11.6% of OI ears, respectively. Multiple linear regression revealed that thresholds progressed by 0.5 dB/year at 0.25 kHz to 0.8 dB/year at 0.8 kHz in the ears with conductive or mixed hearing loss. Pure sensorineural hearing loss progressed by less than 0.1 dB/year at 0.25 kHz to 1.2 dB/year at 8.0 kHz.
The approach to treatment of hearing loss in OI is discussed by Vernick in Pillion et al. . Surgical correction of conductive hearing loss from ossicular fracture and stapes footplate fixation is possible but the results of such surgery are less successful compared to similar surgery in non-OI reports. Stapedectomy results in type I OI show closure of the air-bone gap to within 10 dB in 75%–85% compared to published results in normal patients of 90%–95%. Postoperative hearing loss is also higher in OI patients with up to 8% losing hearing instead of the 1% seen in the non-OI group. Garretsen et al. found a gain in hearing in 85% of 58 ears after 3 months and in 68% of 40 ears followed for 9.6 years and no loss of their postoperative gain in hearing .
Treatment for sensorineural hearing loss in people with OI is similar to people without OI. Hearing aids can help manage sensorineural, conductive and mixed (both sensorineural and conductive) hearing. Fitting a person with OI with a hearing aid is the same as fitting someone without OI . However, hearing aids will not be useful in all OI patients. In that situation, cochlear implants may be useful. Cochlear implants are surgically implanted into the inner ear and electrically stimulate the auditory nerve directly. In OI the operation can be more difficult because of the hypervascularity of the bone and the possible narrowing of the cochlear channel. Also, the lower bone density in OI does not shield the electrical stimulating current which may lead to facial nerve stimulation. Vernick suggests that cochlear implants may provide considerable benefit to deaf individuals with OI. Bone-anchored hearing aid (BAHA) is an alternative treatment for conductive hearing loss and single-sided deafness . It requires that a titanium implants osseointegrate into the bone. A hearing aid is then attached to the implant to stimulate the bone directly. Implantable hearing aids are devices attached to the ossicles surgically that directly drive the ossicular chain. However, the frailty of the ossicles in OI may limit the use of the implantable devices.
48.9.4
Cardiac lesions in osteogenesis imperfecta
Clinically significant cardiac lesions are relatively infrequent in OI, in contrast to other of the heritable disorders of connective tissue such as MFS and EDS.
However, type I collagen, the main structural protein of bone, is also a significant component of myocardium, heart valves, the aortic root and the walls of blood vessels. Thus having either less type I collagen or a structurally defective molecule in cardiovascular tissue may be expected to alter cardiovascular function. This is illustrated by studies of thoracic aorta in the 3-month-old oim/oim and oim /+ heterozygous mouse model of OI. Circumferential biomechanics of oim/oim and heterozygote descending thoracic aortas demonstrated the anticipated reduced Fmax and IEM relative to wild-type mice. Histological analyses of oim descending aortas demonstrated reduced collagen staining and deceased hydroxyproline relative to wild type aortas suggesting decreased collagen content .
Vetter et al. have reported cardiac findings in 58 children aged 1–16 years with different OI types . Congenital cardiac malformations included valvular aortic stenosis with atrial septal defect II (Fallot Tetralogy), holosystolic mitral valve prolapse, and regurgitation. Children suffering from type III OI showed aortic root dilatation (28%) and increased septal (40%) and posterior left ventricular wall thickening (68%) on initial evaluation. Valvular cardiac lesions have been described in 109 individuals with various nonlethal OI syndromes from 66 separate families . Clinically discernible valvular dysfunction was encountered in 4 of the 109 individuals (aortic regurgitation in 2, aortic stenosis in 1, and mitral valve prolapse in 1) none of whom were related. Aortic root dilatation was recognized in eight (12.1%) individuals. In the same cohort, mitral valve prolapse was encountered in 6.9% of a subset, 29 individuals aged 15 years or older .
Thiele et al. reported on 46 children and young adults, aged 3–23 years, with types III and IV OI: 78% of participants had one or more valvular or cardiac chamber findings . Mild tricuspid regurgitation was the most common finding and occurred in over half of the participants with each type. Two type III and 10 type IV patients had combined mild mitral, pulmonic, or aortic regurgitation in addition to the tricuspid valve finding. Seven participants had ECG findings including sinus tachycardia with Q waves.
Echocardiographic studies in Norway involving 99 adults with OI types I, III, and IV, and 52 controls revealed the following: 37% had hypertension, left ventricular end-diastolic internal dimensions (LVIDd) and left ventricular mass were significantly larger in the OI group when compared with the controls, type III OI showed significantly enlarged LVIDd as compared with types I and IV. All aortic diameters were significantly larger in the OI group than in the control group, and in type III compared with types I and IV. Aortic regurgitation was mild in 10.1% of patients, and moderate in 10.1%. Moderate mitral regurgitation occurred in 7.1% in the OI group. By contrast, 38 of 40 OI individuals (95%) studied by Migliaccio et al. showed valvular regurgitation compared to one in control subjects . Ten OI patients (25%) showed mitral regurgitation, 4 had mitral and aortic regurgitation; 12 had mitral and tricuspid regurgitation; and 12 had mitral, tricuspid, and aortic regurgitation. However, all valvular regurgitations in OI patients grade 1/4 and were not hemodynamically significant. About 7.5% had mitral valve prolapse. Diastolic dysfunction was documented in 95% of OI patients and 2% of controls. Although diastolic dysfunction is common in OI adults, the clinical expression of the defect is currently undefined.
Right-sided heart failure may complicate progressive pulmonary insufficiency in type II OI, in type III disease in infants, and also in type III disease in adults .
Animal studies confirm cardiac changes in the presence of type I collagen mutations. Compared with wild-type littermates, homozygous murine oim / oim hearts exhibited 35% lower collagen area fraction, 38% lower collagen fiber number density, and 42% smaller collagen fiber diameter . In the oim , heterozygote descending thoracic aortas demonstrated reduced Fmax and IEM relative to wild type mice. Also, histological analyses of oim/oim descending aortas demonstrated reduced collagen staining relative to wild-type .
The Aga2 mouse model has a dominant frameshift mutation in the Col1(a-1) C-propeptide domain. Cardiac studies in this model (Aga2) severe showed enlarged septa and right ventricular hypertrophy. Matrix collagen in the myocardium was disordered with fewer and thinner collagen fibrils. Ultrasound demonstrated that left ventricular end-systolic internal diameter (LVESD) was significantly higher than control resulting in a significantly lower ejection fraction, suggesting impaired muscle contraction .
The extent of peripheral vascular disease in adults with OI is not known. Carotid artery dissection has been reported in three type I patients . A type I collagen mutation was reported in one patient with carotid artery dissection . The OI Program at the Kennedy Krieger Institute recently encountered spontaneous carotid dissection in four patients, three type I and one type IV individuals. No relation to type I collagen mutations was found (Shapiro, personal communication).
48.9.5
Pulmonary involvement in osteogenesis imperfecta
Pulmonary insufficiency is a major problem for two groups of OI subjects: (1) those neonates with severe or lethal disease and (2) adult types III, IV, severe type V, and types VI and VII disease including those with severe scoliosis. Experience suggests that compromised pulmonary function may occur with relatively small degrees of scoliosis. A restrictive thoracic disease may be complicated by the presence of pectus carinatum or pectus excavatum deformities.
It is common for infants with lethal perinatal disease to succumb during the first few weeks of life from pulmonary insufficiency with superimposed pulmonary infection. Neonatal pulmonary insufficiency may be secondary to the presence of intrauterine rib fractures (e.g., type IIA OI with beading of the ribs), giving rise to dyssynergic state of the thoracic musculature. Tracheomalacia may compromise airway function. Pulmonary hypoplasia has been occasionally reported in postmortem examinations of infants with type II disease. A case of type II OI with pulmonary hypoplasia was studied to determine the potential effect of a mutation affecting the synthesis of pro-α 1 (I) collagen on bronchoalveolar development. In this case, it was determined that the development of the bronchoalveolar tree had ceased at about the 10th week of gestation .
Another cause of pulmonary insufficiency in severe type III OI is alveolar hypoventilation as a consequence of compression of the brainstem secondary to basilar invagination . The type III OI subject may have a restrictive pulmonary disorder secondary to severe scoliosis with a decreased volume of the thoracic cage ( Fig. 48.4 ). Deformity of the chest may increase as the scoliosis worsens: both tend to increase with age leading to dyspnea with little effort. It was initially observed that pulmonary insufficiency in severe OI correlated with the presence of kyphoscoliosis .
Pulmonary findings were reported by Thiele et al. in 46 children and young adults with types III and IV OI . About 78.3% had developed scoliosis greater than 10 degrees (mean g 25 degrees, range 0–70 degrees). With worsening scoliosis, OI patients had a progressive decline of forced vital capacity (FVC), tidal lung capacity (TLC), and vital capacity (VC). Pulmonary function parameters were observed to drop abruptly in patients after 30 degrees of curvature; thereafter, the decline was gradual. Pulmonary function parameters declined significantly with age for all OI patients from nearly normal at age 4 to about half of predicted values by age 20 years. The age-related decline of FVC, TLC, and VC was significantly greater for type III than the milder type IV OI patients.
Using standard spirometry, assessment of rib cage geometry, breathing patterns and changes in regional chest wall volume, Lomauro et al. found OI type III and IV patients to have reduced FVC and forced expiratory volume in 1 second (FEV 1 ) compared to controls. In both seated and supine positions, ventilation was lower in OI patients than controls because of lower tidal volume. Chest wall deformity altered pulmonary dynamics in the type III patients more than in the type IV individuals: pectus carinatum, paradoxical inspiratory inward motion of the pulmonary rib cage, significant thoracoabdominal asynchronies, and rib cage distortions in supine position characterized the type III patients .
We have evaluated sleep patterns with reference to sleep apnea in subjects with type III disease. The results indicate that despite the presence of type III malocclusion of the jaw and displacement of the tongue in certain patients, sleep apnea was found in only two of six type III subjects. Obstructive sleep apnea secondary to laryngomalacia is reported in an adult patient with OI . The treatment of these individuals is complicated by facial asymmetry and their small thoracic volume. Mechanical ventilatory support including the administration of oxygen by bi-level positive air pressure (BiPaP) or continuous positive airway pressure (CPAP) at home is frequently required. In an effort to maximize therapy, it is recommended that pulmonary function be evaluated at 3-year intervals when the patient is stable and yearly when pulmonary insufficiency if present.
48.9.6
Neurologic disorders in osteogenesis imperfecta
A variety of neurologic lesions have been reported in OI patients. The majority involve individuals with moderate-to-severe skeletal deformity. In this category are hydrocephalus, basilar invagination with brainstem compression, basilar impression, and platybasia which may be associated with cortical atrophy . Basilar invagination allows upward displacement of the upper cervical spine and clivus into the foramen magnum. Brain stem compression may result in compression of the upper cord, with sensory neuropathy as well as impaired respiratory function. Syringomyelia has been reported in the presence of basilar impression .
In a review of neurosurgical complications in 10 OI children, Sasaki-Adams et al. observed macrocephaly in five patients, subdural hematoma in three patients, epidural hematoma in two patients, and hydrocephalus in three patients. Basilar invagination and spinal fractures were observed in 20% of the cohort . Charnas and Marini have reported on neurologic disease in 76 OI children, mean age 8 years, the majority with types III and IV disease . Ten patients had macrocephaly, although head circumference was generally normal. Cerebral atrophy was identified in 17 individuals ranging from 7 to 17 years. Eight subjects, the majority with type III disease, had basilar invagination. Seizures occurred in five patients. Ten subjects had suffered skull fractures. Ventriculomegaly consistent with communicating hydrocephalus occurred in 17 patients .
Signs of basilar invagination include headache (76%), lower cranial nerve dysfunction (68%), hyperreflexia (56%), quadriparesis (48%), ataxia (32%), nystagmus (28%), and scoliosis (20%). Four patients (16%) were asymptomatic . A chronic headache syndrome, perhaps due to increased intracranial pressure, occurs in individuals with type III OI where the base of the calvarium is deformed. Trigeminal neuralgia may accompany this syndrome. Paresthesias may occur .
Arponen et al. analyzed lateral skull radiographs and mid-sagittal MRIs of 76 patients aged 0–39 years with either type I, III, or IV OI. The material included a longitudinal series of 31 patients. Craniocervical anomalies were observed in 37% of patients and in all OI types studied. Of the three types of anomalies, basilar invagination was seen in 13%, basilar impression in 15%, and platybasia in 29% of the patients. About 44% displayed more than one type of anomaly .
An analysis of cephalometric abnormalities in OI types I, III, and IV ( n =169) by Cheung et al. showed that height z-score, but not the type of collagen type I mutation, was independently associated with the prevalence of skull base abnormalities . About 48% of patients with a height z-score below −3 had a skull base abnormality regardless of whether or not they had received bisphosphonate treatment as infants. A second category of neurologic complications secondary to skeletal deformities occurs in individuals with mild-to-moderate disease. In this category are nerve entrapment syndromes following fracture healing and nerve root lesions secondary to scoliosis or vertebral collapse. Surgical decompression of the spinal cord may be required in severely affected subjects . There are divergent views regarding the surgical approach to basilar invagination in OI. Traditional approaches to the craniocervical junction include the transoral-transpharyngeal, transmandibular, circumglossal, and transcervical endoscopic routes. A sublabial approach has also been successfully applied . The current methods may include halo traction and anterior or posterior fixation .
48.9.7
Scoliosis in osteogenesis imperfecta
The incidence of spinal curvature approaches 70%. Contributing to the development of scoliosis are vertebral defects secondary to osteoporosis and laxity of the spinal ligaments. In general, the more severe types of OI are associated with greater deformity of the spine. A study of vertebral compression in 46 children with OI demonstrated that vertebral compression began shortly after birth in types III and IV OI and progressed to puberty. The pediatric OI spine was unusual due to the presence of posterior compression fractures, particularly at the L4 and L5 levels . Additional deformities include platyspondyly, thoracic kyphosis, and increased lumbar lordosis. A survey of 102 patients revealed that curvature was mild (<40 degrees) in 50%, moderate (<60 degrees) in 6%, and severe (>80 degrees) in 2% . Deformities of the thoracic cage are almost always present in subjects whose scoliosis is greater than 40 degrees. Clinical records and spinal radiographs of 19 OI patients, average age 14.2 years (range, 4–20 years), were reviewed by Watanabe et al. Seven patients were Sillence type I and 12 patients were type III. The average Cobb angle of scoliosis was 25.2 degrees (range, 5–108 degrees) including five type III patients with an angle of 30 degrees or more. A correlation of scoliosis with the BMD z-score suggested that the pathology of scoliosis was related to vertebral fragility .
Thus scoliosis tends to be mild in type I OI, more marked in type IV disease, but of major concern in type III disease since (1) progression of the deformity may occur with age, leading to compromised pulmonary function, and (2) surgical stabilization may be complicated by the osteoporotic quality of bone that limits effective placement of a prosthesis such as the Harrington rod. As in other forms of scoliosis, exaggeration of the deformity occurs after the age of 5 years and may increase at the time of pubertal growth spurt. As a general rule, progression of scoliosis will occur in most patients. Bracing is of little value in this circumstance and it may further compromise pulmonary function.
48.9.8
Hyperplastic callus formation
This is a very uncommon complication that appears during the healing phase of fractures, including areas that have previously fractured and healed without incident . Any clinical type including type I OI may be affected. However, HPC is a characteristic feature of type V OI . A painful tumor-like inflamed excessive deposition of callus forms from ECM. HPC is frequently misdiagnosed as osteosarcoma. Osteosarcoma is a consideration in the differential diagnosis of localized pain and swelling, but osteosarcoma is extremely rare in OI patients. Callus tissues in OI have been studied without finding a significant deviation from the normal pattern of fracture healing aside from overmodification of types I and III collagen . The course of HPC is gradual resolution, usually over a period of several weeks to months. Recurrence has been observed in a few patients. Treatment may include administration of glucocorticoids to suppress matrix formation and the apparent inflammatory component of this reactive lesion. Neither pamidronate nor indomethacin was found to alter the course of HPC . Low-dose radiation therapy has been reported to be successful in decreasing pain and swelling associated with HPC formation .
48.9.9
Nonaccidental injury
The diagnosis of OI is frequently raised during legal proceedings for nonaccidental injury (NAI). This differentiation should be based on clinical grounds . A review of biochemical testing completed on cells from 262 infants with OI versus infants with unexplained fractures described three children who were not previously identified as OI by comprehensive clinical evaluation . Thus DNA analysis should be requested during these evaluations. Depending on the child’s phenotype it may be necessary to request not only COL1A1/A2 gene sequencing but also sequencing the recessively inherited genes. However, it is recognized that all infants with recessive OI have obvious radiographic abnormalities fitting a diagnosis of OI and not of NAI .
48.9.10
Medical treatment in osteogenesis imperfecta
Children and adults with OI should have adequate calcium intake as currently recommended in the Dietary Reference Intakes of the US Department of Agriculture. Because urinary calcium may be elevated in some children, urinary calcium should be measured if a significant increase in dietary calcium is prescribed.
Approximately 50% of children and adults with OI will be found to be vitamin D insufficient (serum levels <32 ng/mL) on initial testing. In a cross-sectional study of 315 OI patients aged 1–17 years, with OI types I, III, and IV, Edouard et al. found a positive correlation between serum vitamin D levels and lumbar spine BMD z-scores . However, using bone histomorphometry, these authors did not find a relationship between serum vitamin D levels and bone mineralization and bone mass in children between the ages of 1.4 and 17.5 years .
Bisphosphonates, administered orally or intravenously, have been considered the mainstay of treatment for children and adults with OI. Although there are multiple observational studies reporting a decrease in fracture rate in children with OI, after a decade of experience, qualifications regarding bisphosphonate treatment have surfaced both as regards the effectiveness of different bisphosphonates as well as the uniformity of response in children and adults.
The current status of cyclical treatment with pamidronate in OI children has been summarized by glorieux: intravenous pamidronate administration will increase bone mass, reduce bone pain, increase the size of vertebral bodies, and decrease fracture incidence by approximately 50%. There is improved ambulation secondary to a decrease in musculoskeletal pain . However, Marini has cautioned that although bisphosphonates increase BMD, whether they also improve fracture rates or functions of daily life is unclear .
To date, two critical reviews of bisphosphonate use in children with OI have been published. A Cochrane review of eight randomized trials in children including 403 participants reported a significant reduction in the number of fractures after bisphosphonates in only one of these trials: no differences in fracture rate were reported in three other trials . Considering pamidronate versus placebo, two studies showed differences in the number of participants experiencing at least one fracture, and another study showed no difference in fracture incidence. Thus it was not clear that either oral or intravenous bisphosphonates uniformly decreased fracture rate. Castillo and Samson-Fang also identified eight studies in children that confirmed improvement in bone density and found a 30%–60% reduction in fracture risk in three of four small randomized controlled trials . Whereas intravenous pamidronate may decrease fracture rate in children, two recent studies have demonstrated that oral bisphosphonates, risedronate and alendronate, will not decrease fracture rate in children with OI .
Clinical trials are in progress in children with OI using zoledronic acid, a third-generation amino bisphosphonate that exhibits a long duration of action. This drug has the advantage of administration over a shorter time interval compared to pamidronate. However, zoledronic acid is more powerful compared to pamidronate so that its effect on suppression of bone turnover remains a question and appropriate dosing and treatment schedules remain to be defined.
As yet unanswered are questions related to the indications for treatment, the frequency of pamidronate infusions and the duration of treatment . The current recommendations are that treatment be considered in children with moderate-to-severe OI, in those children with recurrent extremity fractures, vertebral compression fractures, and reduced bone mass .
The impact of bisphosphonate treatment on bone turnover and the duration of treatment are important to assess in the individual patient. Rauch et al. evaluated histologic changes in bone biopsies following more than 4 years of pamidronate treatment in children aged 1.4–15.3 years at baseline. DXA BMD increased by 72% in the first half of the observation period, but by only 24% in the second half. Iliac crest biopsy showed that mean cortical width and cancellous bone volume increased by 87% and 38%, respectively, between baseline and the first time point during treatment. Cortical width did not change significantly thereafter but there was a trend ( P =.06) toward higher cancellous bone volume. The average bone formation rate on trabecular surfaces decreased by 70% after pamidronate treatment and showed a trend ( P =.08) toward a further decline in the second part of the study interval. These results are significant in that the response of bone achieved with pamidronate treatment appears to largely occur during the first 2–4 years .
In contrast to the positive results in children, in adults with OI, bisphosphonate treatment, with either pamidronate or oral bisphosphonates, will increase bone density but will not decrease fracture incidence except to a marginal extent in individuals with type III and IV OI . In a result similar to that in children, treatment of adults with oral risedronate increased BMD at the lumbar spine but not at the total hip, decreased bone turnover but did not decrease fracture rate . Thus, as of this date, it is not advised that adults with OI receive treatment with bisphosphonates.
Because of the frequent occurrence of short stature in OI children, growth hormone (GH) treatment has been used in children with OI. In general, baseline provocative tests of GH and insulin-like growth factor (IGF)-I have been normal in OI subjects although a blunted somatomedin generation test response to GH was observed in 13 of 22 children . GH was found to increase the rate of linear growth velocity, increase bone turnover and mineral content in lumbar trabecular bone in patients with collagen defects typical of mild/moderate OI .
Marini administered recombinant GH (rGH) to 26 children with types III and IV OI, aged 4.5–12 years . Children were treated with rGH, 0.1–0.2 IU/kg/day for 6 days/week, for at least 1 year. Approximately one-half of the treated OI children sustained a 50% or more increase in annual linear growth over their baseline growth rate. Most responders (10 of 14) had moderate type IV OI: other types showed less response. Responders, largely in the type IV patients, were distinguished from nonresponders by higher baseline PICP values suggesting that they had an intrinsically higher capacity for collagen production. GH has not been accepted as a primary treatment agent for OI children because the gains in height are limited to certain phenotypes and because the effect on fracture rate is not well established. There are also the issues of potential side effects: a GH-induced increase in muscle strength could adversely affect weak bone. GH treatment is expensive. GH treatment in adults is not indicated unless GH deficiency has been confirmed.
The question of estrogen replacement therapy in postmenopausal women with OI is unresolved. It is assumed that the negative effect of estrogen deficiency on bone might be similar to that in non-OI individuals but this has not been studied.
Potential pharmacological treatments for OI involve: receptor activator of nuclear factor kappaB ligand (RANKL) inhibitors, cathepsin K, and sclerostin inhibitors. Teriparatide (human recombinant parathyroid hormone 1–34) is currently in clinical trial. Molecular strategies include hammerhead ribozymes and small interfering RNA methodology. Investigational methods involve gene targeting using adenovirus vector (AAV) to inactivate mutant COL1A1 genes and allogenic mesenchymal cell engraftment via intrauterine fetal transplantation which is reported to decrease fracture rate in newborn animals . Stem cell therapy involving induced pluripotent stem cells as a means of repopulating the marrow with functional osteoblasts producing normal type I collagen may offer great therapeutic potential.
48.9.11
Rehabilitation and physical therapy
Montpetit et al. have surveyed 24 patients (mean age: 25.0 years) with OI types I, III, IV, and V. Participants with OI type I reported full independence, and only few respondents with OI types IV and V reported some limitations in mobility and domestic life activities. Young adults with OI type III had significantly lower activity scores in aspects of mobility and domestic life and lower levels of participation in employment, sporting activities, and transportation .
Consistent and coordinated rehabilitative and physical therapies are critical to the successful development of children with OI and to the maintenance of effective daily activities for adults with this disorder. Functional independence is the ultimate goal. Binder et al. have stated the two central tenets in approaching rehabilitative care: (1) the variability associated with the OI phenotypes makes it difficult to predict which children are at risk for significant disability and (2) one must fully evaluate a child’s functional abilities and potential for rehabilitation .
Adults with severe disease may be unable to sit independently, and many adults with type III disease are wheelchair dependent or, as with type IV subjects, dependent on lower extremity braces, canes, or crutches for ambulation. Important issues involve the development and maintenance of muscle strength in the upper and lower extremities, the prevention of joint contractures at several joints including the shoulders, hands, hips, and feet; poor joint alignment; and disturbances of gait and low endurance in those who ambulate. To approach optimal rehabilitation and maintenance of function, children were classified as to the severity of disease and potential for rehabilitation and a specific program was outlined for each group . Modalities included custom-molded seats to align hips, knees, and ankles to prevent stress in the spine, therapeutic water activities, soft tissue mobilization techniques to control joint contractures, and specific exercises to strengthen muscle groups, improve posture and balance, and increase endurance. Specific bracing techniques involving both joint support and lightweight lower extremity braces were employed to limit joint instability and permit early ambulation. As a result, improvements were found in head and trunk control, joint alignment, strength in the extremities, and the ability to ambulate.
48.9.12
Orthopedic treatment in osteogenesis imperfecta
Orthopedic management of OI starts literally at the moment that the diagnosis is made. In the infant, this may involve aligning fractures to minimize deformities and rodding bones to decrease the occurrence of fractures and improve function . Intramedullary rodding is, in the author’s opinion, indicated as early as required to decrease the risk of fracture and improve function. Both nonexpanding and expanding rods are used, depending on the age of the child, the size of the bone, and the severity of the osteopenia. Expanding (Bailey–Dubow, Fassier–Duval) rods can elongate with the growth of the bone. However, these rods may be associated with bending, unscrewing of the T-shaped end from the sleeve of the rod, and breakage . These rods may fail to elongate and the T-end of the rod may migrate within the bone. Protrusion through the bone may occur ( Fig. 48.10 ). Overall, complications occur in about one-third of cases and these require reoperation in approximately 15%–20% of cases. Birke et al. have reported on the recently developed Fassier–Duval rod in the OI patient group . Anchored at both ends, this rod was associated with a 13% reoperation rate for proximal rod migration and a 40% complication rate: rod migration and limited telescoping, and intraoperative joint intrusion.
Scoliosis in OI is discussed earlier (pulmonary). Correction of scoliosis will decrease the progressive loss of pulmonary function. Placement of Harrington rods and fusion for progressive scoliosis is a major orthopedic concern. The therapeutic result may be complicated by the quality of the bone into which a prosthesis is seated, the degree of scoliosis, and the age of the patient relative to the growth rate.
In the teenager or adult, orthopedic care is required to assess and surgically correct scoliosis, stabilize lax joints with equalization of the length of extremities, and correct deformity by osteotomy. Protrusio acetabuli, a source of chronic pain and limited mobility, has been reported in 29% of subjects with type III OI . Joint replacement of the hip and knee has been performed with satisfactory results for five of six patients having the following complications of OI: osteoarthrosis of the hip and knee, severe deformity of the hip associated with posttraumatic arthritis, acetabular fracture, and nonunion of a subtrochanteric fracture .
48.10
Osteoporosis in the heritable disorders of connective tissue
48.10.1
Homocystinuria as a cause of adult osteoporosis
Homocystinuria (OMIM 236,200), an autosomal-recessive disorder of connective tissue, is associated with mental retardation, ectopia lentis, marfanoid habitus, and thrombotic vascular disease that occur at an early age. Premature osteoporosis in association with other skeletal alterations occurs in teenagers and may be associated with vertebral and appendicular fractures . Additional skeletal findings include the development of scoliosis, increased length of long bones and growth arrest lines, bowing and fracture of long bones, arachnodactyly, enlarged carpal bones and pectus excavatum, and carinatum deformities of the sternum. Homocysteine is an intermediate in the transsulfuration pathway that converts methionine to cysteine and ultimately to sulfate . The occurrence of homocysteine in urine may result from seven different genetic abnormalities, the most common of which is cystathionine p -synthetase (CBS) deficiency . Although at least 150 different mutations in the CBS gene have been identified since this deficiency was established in 1964, mutations involving c. 833T>C (p. I278T) are present most frequently .
CBS deficiency is inherited as an autosomal-recessive trait . These subjects accumulate abnormal amounts of homocysteine with total plasma homocysteine above 100 μmol/L. Methionine is elevated in plasma due to remethylation of the elevated homocysteine. Other inborn errors similar to that in vitamin B 12 (cobalamin) deficiency affect the remethylation pathway of homocysteine back to methionine . Serum methionine levels are elevated in cystathionine b -synthase deficiency but are low or normal when 5-methyltetrahydrofolate-dependent homocysteine methylation is diminished. Although significant progress has been made in understanding the relationship between elevated plasma homocysteine and premature atherosclerosis in this disorder, little has occurred toward understanding the development of osteopenia at an early age in this syndrome.
Osteoporosis is one of the more common and consistent manifestations of elevated serum levels in homocystinuric subjects. In a series of 26 subjects, 25 were found to be osteoporotic . Approximately 50% of affected individuals will be osteoporotic by the start of their third decade. Vertebral bodies appear osteoporotic, flatter than normal, and elongated in the anteroposterior axis with a posteriorly placed biconcave deformity similar to that seen in hemolytic disorders . Skovby et al. have observed that the finding of individuals with homozygous CBS mutations falls far short of the expected incidence: such individuals appear clinically unaffected or are ascertained with only thromboembolic events as young adults . This suggests that plasma homocysteine should be measured in young adults presenting with osteoporosis.
A quandary related to the frequent appearance and early onset of osteoporosis is the relationship of elevated plasma homocysteine to defective collagen synthesis and, ultimately, to bone loss. This question remains unresolved in spite of early studies suggesting an effect of homocysteine, in vitro, on collagen cross-linking, as demonstrated by increased skin collagen solubility and the failure of collagen to form a stable gel after heating to 37°C and cooling . Homocysteine at concentrations of levels found in patients’ sera will not inhibit lysyl oxidase activity but will prevent the formation of insoluble fibrils and bifunctional cross-links . The assembly of fibrillin-1 in connective tissue matrix is dependent on fibronectin. Studies have suggested that homocystinylation of fibronectin will interfere with the fibronectin/fibrillin-1 interaction and will compromise fibrillin-1 deposition in the ECM .
Several reports have documented an association between mild homocysteinemia and premature vascular disease. These subjects are heterozygotes for CBS deficiency . It is not known whether these individuals are also at risk for osteoporosis. The treatment of patients with CBS deficiency includes administration of vitamin B 6 , low methionine diets, and, recently, betaine. This agent will lower plasma homocysteine by increasing the methylation of homocysteine to methionine . The effect of treatment on bone mass is not reported.
48.10.2
Adult hypophosphatasia
Hypophosphatasia (OMIM 171,760) is a rare inherited disorder characterized by low serum ALP activity and defective bone mineralization (osteomalacia) predisposing to poorly healing pseudofractures, clinical fractures, and defective mineralization of teeth . The inheritance pattern is variable in hypophosphatasia, either autosomal-dominant or autosomal-recessive transmission is recognized.
Six clinical forms of hypophosphatasia are recognized: perinatal lethal, infantile, child, adult, and a benign prenatal form detected in utero, which usually confers lethality, but which tends to lessen in severity postnatally . Because of the marked variation in severity in each of these forms, the disorder may be misdiagnosed as OI both in infants and in adults.
Hypophosphatasia is due to mutations involving the liver/bone/kidney ALP gene that encodes the tissue nonspecific ALP. Associated with a reduction in serum ALP, there is increased urinary excretion of phosphoethanolamine. Whyte et al. have reported that autopsy tissue from three affected subjects with infantile hypophosphatasia, which was profoundly low in ALP activity, had essentially normal levels of pyridoxal-5′-phosphate (PLP), pyridoxal, and total vitamin B6 content despite markedly elevated plasma PLP levels (5800; 14,500; and 98,500 nM; adult norm 5–109 nM) . The histologic results demonstrate an unexpectedly high prevalence of mineralization defects, that is, a pathologic increase in osteoid. Indeed, 36.1% of the analyzed patients presented with an osteoid surface per bone surface (OS/BS) of more than 20% .
Considerable variation occurs in the clinical expression of hypophosphatasia. Adult hypophosphatasia may present in middle-age with recurrent stress fractures of the lower limbs as a result of osteomalacia. The dental history should be carefully evaluated for premature dental loss of permanent teeth, severe dental caries, and alveolar bone loss. There is an increased incidence of chondrocalcinosis and osteoarthritis. Improvement in adult hypophosphatasia has been reported following treatment with teriparatide. However, this is not a consistent result in different clinical trials .
48.10.3
Marfan syndrome and Loeys–Dietz syndrome
MFS (OMIM: 154,700) is an autosomal-dominant disorder characterized by skeletal abnormalities, cardiovascular lesions, and ocular defects . MFS occurs with an estimated frequency of 5/10,000 individuals; 20% of the cases occur due to a new mutation. It affects both males and females equally. Decreased BMD is observed in MFS but the reported incidence is variable.
Loeys–Dietz syndrome (LDS; OMIM: 609,192) is distinguished from MFS by the occurrence of hypertelorism, bifid uvula or cleft palate, and arterial tortuosity with widespread vascular aneurysms and a high risk of aortic dissection at an early age. Decreased BMD and fractures occur in adolescents with this disorder . LDS is transmitted as autosomal dominant .
A paradigm shift in understanding MFS and LDS occurred following the discovery of fibrillin-1 gene (FBN1) mutations in MFS . FBN1 is a major component of extracellular microfibrils. Subsequently, mutations involving genes for transforming growth factor-beta receptors 1 and 2 (TGFBR-1, TGFBR2) have been reported in both MFD and LDS .
MFS-1 is considered as the classic MFS related to a mutation in the FBN1 gene. Mutations of the FBN1 gene are associated with aortic dilatation, ectopia lentis and systemic features including scoliosis, arachnodactyly, mitral and aortic valve lesions, and dural ectasia. MFS-2 is phenotypically indistinguishable from the classic MFS but is related to mutations in the TGFBR1 or TGFBR2 genes . LDS has been subdivided into LDS type I (LDSI) and type II (LDSII) on the basis of the presence or the absence of craniofacial involvement, respectively. Furthermore, LDS II patients display at least two of the major signs of vascular EDS .
LDS results from mutations in the TGFBR1 and TGFBR2 genes which alter the transmission of the subcellular TGF-β signal. TGF-β signaling transduction through both the canonical Smad-dependent and noncanonical Smad-independent signaling pathways such as a Wnt pathway alter the ability of the Runx2 gene to control mesenchymal and osteoblast cell differentiation . With respect to involvement of fibrillin-1 and TGF-β signaling in bone, TGF-β is stored in an inactive form bound to latent TGFB binding protein which is, in turn, bound to fibrillin microfibrils. Mutations in the FBN1 gene directly affecting fibrillin microfibrils or proteolytic injury release TGF-β.
There is extensive clinical overlap between patients with TGFBR1 and TGFBR2 mutations and MFS-1, MFS-2, and LDS . However, clinically, LDS exhibits a more aggressive course than MFS or the vascular type IV EDS, with higher morbidity and mortality due to the complications of arterial and aortic dissections . The medical management of aneurysmal disease in MFS and LDS is mainly focused on the use of p-blocking agents that reduce hemodynamic stress of the aortic wall .
A cause-and-effect relationship has been suggested between an alteration of the TGF-β pathway signaling due to TGF-β2 receptor mutations and aortic disease in MFS and LDS . Blockade of angiotensin receptor by losartan has been demonstrated to limit muscle loss in fibrillin-1 deficient mice in which TGF-β signaling is increased . This has been used to advantage in the current treatment of arterial and aneurysmal disease with losartan which blocks the angiotensin II receptor . Angiotensin II type 1 receptor is present in the aorta and is activated by mechanical stress which stimulates cell growth, inflammation, and vasoconstriction .
The role of TGF-β in MFS and LSD aortic disease is based in part on the following: in comparison to healthy controls, immunohistochemical studies of aorta from MFS patients have demonstrated matrix fibril and elastic fiber disruption with enhanced TGF-β1 in the cytoplasm, and enhanced activity of the intracellular Smad signaling pathway, along with attenuated TGF-β1 receptor in the aortic tissue . Thus MSF patients may have dysregulated TGF-β signaling associated with increased collagen deposition and interlamilar elastic fiber degenerative changes in MSF aorta. However, the specific role of TGF-β in aortic disease remains to be fully defined .
BMD in MFS has been reported by several groups and the results are variable raising question as to the extent to which defects in fibrillin-1 or TGF-β signaling affect skeletal tissues. Thirty-two women and 16 children with MFS were studied by Kohlmeier et al. . In the women, mean age 38 years, BMD was reduced at L2–L4, femoral neck, trochanter, and intertrochanter areas compared with age-predicted values. z-Scores for L2–L4 and for the femur neck, trochanter, and intertrochanteric region were −0.59, −1.25, and −1.03, respectively. In MFS children, femoral neck BMG was reduced (z-score=−0.74±1.22) with a nonsignificant trend toward decreased BMD at L2-L4 (z-score=33±1.48).
Ten percent of 60 adult MFS patients (40 women, 20 men), mean ages 32.9±9.3 years, had a history of fracture . z-Score of the hip was −1.26, femur neck −0.93 1.09, and trochanter −1.31. z-Score of the radius (1/3 proximal site) was −1.29. A similar decrease in BMD at the lumbar spine, total hip, and femur neck was observed in 25 OI adults when compared to age- and sex-matched controls . While z-scores were reduced at each site in women, in men BMD was reduced at the femur neck but not significantly lower at the lumbar spine and total hip.
Gasmpietro reported that adult males with MFS demonstrated significantly reduced femoral neck BMD with an average T-score of −1.54 ( P <.001), diagnostic of osteopenia. Although osteopenia and osteoporosis were observed in several middle aged and pre- and postmenopausal women, the average T-score value for adult females and children were within normal limits .
In a series of 130 patients, Moura et al. reported a history of fractures in 24.6%. z-Score values were significantly decreased in the patients at the femoral neck (−1.190±0.098, P <.0001) and wrist (−1.403±1.06; P <.001). Patients had significantly lower BMD values at the femoral neck compared to the height-matched controls (0.841±0.15 vs 1.010±0.017; P <.0001) .
These studies suggest that patients with MFS may have an increased fracture rate, but gender and age distribution is not possible from the reported data. In addition, the reported z- or T-scores are generally in the “osteopenic” range.
BMD values are not reported for LDS at this time. The experience at the Kennedy Krieger Institute and Johns Hopkins Hospital suggests that decreased bone mass is a common finding in adolescents and adults with this syndrome (Tan, personal communication, 2012). Iliac bone histomorphometry in two LSD patients, aged 24 and 17 years, showed elevated bone turnover and mildly low trabecular bone volume per tissue volume. BMDD in trabecular bone was increased in each patient . LDS can be associated with low bone mass and high bone turnover but increased matrix mineralization of trabecular bone.
48.10.4
Ehlers–Danlos syndrome and adult osteoporosis
EDS constitutes a heterogeneous group of connective tissue disorders that have in common joint and skin laxity, excess bruisability, and abnormal wound healing . Osteoporosis occurs in a small number of EDS patients, although this may be underreported. The individual EDS phenotypes vary in the extent of joint laxity and skin fragility, and the expression of other characteristics that clinically identify each variant. Additional manifestations of EDS include recurrent joint dislocations as well as fragile skin with characteristic “cigarette paper” scars, mitral and tricuspid valve prolapse, kyphoscoliosis, and ocular fragility (type VI), and fragile vascular and cavity lining tissues with arterial and gastrointestinal rupture (type IV). Types I, II, and III are dominantly inherited and vary in terms of joint laxity and involvement of the skin. Several syndromes, type V (lysyl oxidase deficient), type VII (short stature, multiple joint dislocations, round facies), type VIII (peridontitis), and type IX (bladder diverticulae, occipital horn syndrome), have unique facies and body habitus that aid in diagnosis.
The EDS types listed earlier had previously been defined based solely on clinical signs. In 1997 a simplified classification was proposed dividing EDS into six major clinical types, including genetic defects were known . This classification grouped EDS types I and II into the classical type , former type III EDS into the hypermobility type , EDS type IV into a vascular type , a kyphoscoliosis type includes EDS VI, former types VII A and VII B are now grouped into an arthrochalasia type.
Several poorly differentiated EDS types are grouped into other forms pending biochemical confirmation of their identity. These include human dermatosparaxis (EDS VIIC), X-linked EDS type V, EDS VIII associated with periodontitis, and EDS type X or fibronectin-deficient EDS associated with prominent bruising. Type IX EDS (occipital horn syndrome), an X-linked recessive disorder, shares biochemical features with the Menkes disease and has been categorized as a disorder of copper metabolism .
Mayer et al. have collated a summary of mutations currently reported in EDS 1-VII . Two recently described EDS-phenotypes involve (1) mutations in the SLC39A13 gene which encodes a membrane-bound zinc transporter (spondylocheirdysplastic form of EDS) and (2) a syndrome of severe scoliosis, skin and joint hyperelasticity, and progressive facial coarsening. This EDS phenotype is associated with mutations involving RIN2 , which encodes Ras and Rab interactor-2 which is involved in endocytosis . RIN2 deficiency was found to be associated with paucity of dermal microfibrils and deficiency of fibulin-5, which may underlie the abnormal skin phenotype displayed by the patients.
A third clinical variant characterized by severe muscle hypotonia at birth, progressive scoliosis, joint hypermobility, hyperelastic skin, myopathy, and sensorineural hearing impairment was reported by Baumann et al. . Clinically, the disorder resembled the kyphoscoliotic type of EDS (EDS VIA) and was caused by either homozygous or compound heterozygous mutations in FKBP14.
The classic EDS types are associated with mutations in the COL5A1 and the COL5A2 genes located respectively on chromosomes 9p and 2q31–32, which encode type V collagen . Type V collagen is a member of the group of fibrillar collagens and is composed of three a polypeptide chains (1(V) 2 -2(V)), which are products of the COL5A1, COL5A2, and COL5A3 genes. In the majority of patients with molecularly defined classic EDS, the disease is caused by a mutation leading to a nonfunctional COL5A1 allele and resulting in haploinsufficiency of type V collagen. A smaller proportion of patients with classic EDS harbor a structural mutation in COL5A1 or COL5A2, causing the production of a functionally defective type V collagen protein .
Tenascins (TNX) are a family of ECM proteins that affect the spacing of collagen fibers . There are four family members: TNX-X, TNX-R, TNX-W, and TNX-C. TNX-X associates with type I collagen, and its absence can cause an autosomal-recessive Ehlers–Danlos phenotype . Clinically, the phenotype resembles the classical type of EDS, but wound healing is normal; atrophic scars are absent. The clinical diagnosis can be confirmed by the absence of TNX in the serum and by mutation analysis of the TNXB gene.
Type IV EDS is the result of different mutations involving the COL3A1 gene which encodes the a chains of type III collagen . The majority of mutations involve point substitutions of arginine, serine, valine, aspartic acid, or glutamic acid for glycine in the triple-helical domain. Small genomic deletions, multiple exon deletions and exon skipping have also been reported in EDS IV .
The group of disorders classified as EDS type VIIA and EDS VIIB are the result of mutations involving the N-terminal a chain propeptide cleavage site . EDS VII C, the homolog of dermatospraxasis in sheep and cattle, is the result of defects in the converting enzyme procollagen N-peptidase and the procollagen N-proteinase ADAMTS2 .
Overlap of the classic EDS phenotype with OI has been reported in individuals with COL1A1 or COL1A2 mutations that interfere with normal N-protease processing of the N-terminal propeptide similar to COL type 111 mutations in type VII A/B EDS .
In the original description of EDS VI (kyphoscoliosis, ocular fragility type), it was found that the hydroxylysine content of collagen in skin and tendons was less than one residue per 1000 total amino acids in contrast to four residues per 1000 in control tissue . The hydroxylysine content was somewhat less than normal in bone and was normal in cartilage. Skin fibroblasts from the majority of patients with EDS type VI type have significantly decreased lysyl hydroxylase (LH) activity due to mutations in the LH1 gene. Both low LH (type EDS VI A) and normal enzyme levels (type EDS VI B) have been reported in fibroblasts obtained from individuals with this form of EDS .
The previously categorized EDS IX is a rare X-linked condition characterized by skeletal dysplasia, characteristic occipital “horns” that appear during adolescence, diarrhea due to increased bowel motility, and obstructive uropathy due to bladder diverticulae that appear during the first decade. This condition is allelic with Menkes disease and along with Wilson disease is one of the three hereditary disorders of copper metabolism. Cells from patients with these disorders have elevated levels of intracellular copper due to defective copper transport. A defective copper transporting ATPase gene ATP7A demonstrated in Menkes syndrome may underlie the mechanism of this disease. The copper transporting P-type ATPase ATPP7B is mutated in subjects with Wilson disease who also may be osteoporotic as children or adults .
Patients with EDS may show vertebral abnormalities including wedged vertebrae and spondylolisthesis. Few data exist on BMD in the various EDS phenotypes. However, Coelho et al. assessed BMD in four patients, aged 16–25 years, with EDS type I. Bone density at the lumbar spine was persistently 1 standard deviation (SD) below average for age and sex. However, this difference was not present for the femur neck .
Yen et al. have reported skeletal findings in Formosan EDS patients, aged 13 months to 36 years. BMD studies performed in 11 patients revealed that all had osteoporosis. Bone fracture(s) had occurred in three of 16 patients .
In the context of adult osteoporosis, diagnostic uncertainty involves subjects with IOP in whom joint laxity is of moderate degree and the question of mild EDS arises. Mutation identification is advised where the phenotype is not well defined. Certain individuals with N-terminal COL1 mutations may have features of both OI and EDS .
48.10.5
Idiopathic juvenile osteoporosis
Idiopathic juvenile osteoporosis (IJO) is included in this section because this is considered along with mild OI in the differential diagnosis of idiopathic osteopenia in teenagers and young adults. IJO is an uncommon self-limited disorder of children and teenagers characterized by potentially reversible osteoporosis that usually appears in the prepubertal years. Norman found approximately 60 cases of IJO reported in the literature between 1939 and 1991 . There is no gender selection. Although differentiation from type I OI may be difficult, IJO is not familial and not associated with blue sclerae, DI, or short stature. Certain patients may have a pectus carinatum chest deformity.
The disorder usually has its onset 2 or 3 years before puberty, although the age at onset may vary from 3 to 16 years . It usually runs its course over 2–4 years. Children complain of the gradual onset of pain in the back, knees, and ankles. Wedge compression fractures of the spine and lower extremities may occur, causing kyphoscoliosis, as may fractures of the knees and ankles. Although bone loss eventually ceases and remineralization proceeds, mild cases are left with short stature and mild kyphosis. In severe cases, IJO may lead to marked deformities of the extremities and pulmonary insufficiency due to kyphoscoliosis and collapse of their rib cage .
Serum biochemistries are normal in IJO. Calcium balance may be negative during the period of rapid bone loss. Urine calcium excretion has been reported to be normal or increased . Bone biopsies have revealed evidence of either increased bone resorption or normal resorption and decreased bone formation . Rauch et al. examined iliac crest bone biopsies in six children with IJO, aged 9–12 years, compared to healthy children. Intracortical bone remodeling and BS metabolic parameters (osteoid, osteoblast, and mineralizing surface and bone formation rate) were similar in IJO and controls. Decreased remodeling activity on the endocortical surface of the internal bone cortex was identified .
Radiologic examination in IJO reveals generalized osteopenia associated with a decrease in height of vertebral bodies due to wedge-shaped fracture or misshapen vertebral bodies due to collapse of the endplates . Long bones are osteopenic and may show osteoporosis of newly formed bone during the pubertal growth spurt (neo-osseous osteoporosis). Linear metaphyseal rarefaction is a clue to this disorder. It results from impaction type fractures that occur at the growing ends of weight-bearing bones . These fractures are typically seen at the distal tibia adjacent to ankle joint and distal femur adjacent to the knee.
Treatment in children with IJO should depend on the presence of low bone density by DXA as well as a fracture history involving long bone and vertebrae .
48.10.6
Idiopathic osteoporosis in young adults
IOP refers to males under the age of 50 years and premenopausal females (see Chapter 45 : Glucocorticoid-Induced Osteoporosis and Cushing’s Syndrome). Both the incidence of this disorder and the mechanisms leading to diminished bone mass and fractures in this population are unknown. A family history of osteoporosis is common. In a cohort of 61 females with presumed IOP, secondary causes (amenorrhea, anorexia nervosa, glucocorticoid exposure) were present in 43% while 39% had IOP .
The syndrome is characterized by low bone density and fragility fractures although the diagnosis of IOP does not require the occurrence of fracture . Studies in both males and females have shown the heterogeneity associated with IOP both with regard to bone turnover and bone architecture. In males, the spine is the predominant site of bone loss with either cortical or trabecular bone loss occurring in individual patients .
Bone biopsies in males with IOP show low rates of bone turnover with low mineral apposition rates and decreased trabecular bone formation . Serum IGF-I levels are low in males with IOP . In women with IOP bone turnover rates may be either high or low. Individuals with high bone turnover seen on iliac crest bone biopsies tend to have elevated levels of osteoclast-related proteins (TRAP5b) and parathyroid hormone and higher urine calcium excretion than controls. However, IGF-I levels are normal in women with IOP .
The phenotype of IOP resembles that of type I OI in many patients. However, in IOP fractures start later in life, frequently above age 20 years, as compared to OI where there are childhood fractures. In these patients, diagnostic studies, if warranted by fracture history, may be limited to COL1A1 and COL1A2 genes. The treatment of IOP in young adults, with either an anabolic agent such as teriparatide or an antiresorptive bisphosphonate, is not currently defined.
References
Related posts:




Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


