Oncogenic Viruses and Tumor Viruses
6.1 INTRODUCTION
Viruses are implicated in approximately 15% to 20% of all cancers (reviewed by Nevins, 2007; Ou and Yen, 2010). They can cause malignancies that include nasopharyngeal carcinoma, Burkitt lymphoma, cervical carcinoma, T-cell leukemia/lymphoma, hepatocellular carcinoma, Merkel cell carcinoma, and Kaposi sarcoma. Even more importantly, oncogenes and tumor-suppressor proteins were first identified through the study of cancer-causing viruses. For example, research with simian virus 40 led to the discovery of tumor-suppressor genes, p53 and the retinoblastoma gene (Rb). Oncogenic viruses fall into 2 groups: the DNA tumor viruses that contain either linear or circular double-stranded DNA and the RNA-containing tumor viruses (also called retroviruses). DNA tumor viruses usually cause malignant transformation by inhibiting the normal function (growth control) of tumor-suppressor genes (see Chap. 7, Sec. 7.6), whereas retroviruses usually deregulate signal transduction pathways (see Chap. 8).
Stehelin et al (1976) demonstrated that Rous sarcoma virus (a retrovirus that causes sarcomas in chickens) contained nucleotide sequences that were not found in similar non-transforming retroviruses. These novel retroviral sequences, however, were closely related to nucleotide sequences present in the DNA of normal chickens. This important discovery indicated that a viral transforming gene (in this case v-src) was derived from a normal cellular gene. Many other retroviruses have been studied since and have been shown to contain different oncogenes derived from and closely related to their cellular counterparts. The normal cellular genes from which the retroviral oncogenes (v-onc) are derived are referred to as protooncogenes (or c-onc). The process by which protooncogenes become integrated into the viral genome and are converted to viral oncogenes with overt transforming activity is complex; it involves recombination between the retroviral and cellular genomes following integration of a retrovirus adjacent to a cellular protooncogene. This process, known as transduction, is accompanied by alterations in the structure and regulation of oncogene sequences. Many of the oncogenes found in transforming retroviruses have also been identified independently in spontaneously arising tumors of nonviral origin, where they appear to be activated by other mechanisms, including point mutation, gene amplification, and chromosomal translocation.
Protooncogenes encode a wide range of protein products involved in the control of cell proliferation and differentiation, including growth factors, growth factor receptors, components of signal transduction pathways, and transcription factors that regulate the synthesis of messenger RNA (mRNA). Tumor-suppressor genes, in contrast, represent genes that are likely to play a role in negatively regulating cell growth. In this chapter, the mechanisms of cellular transformation by oncogenic viruses are described. These mechanisms provide clues to more general mechanisms of transformation caused by increases in dominantly acting oncogenes or inactivation of tumor-suppressor genes.
6.2 DNA TUMOR VIRUSES
6.2.1 Polyomaviruses
The polyomaviruses include simian virus 40 (SV40), mouse polyomavirus, and 8 human viruses, JC virus, BK virus, KI virus, WU virus, HPyV6, HPyV7, HPyV9, and Merkel cell carcinoma virus (van Ghelue et al, 2012). Presently, genome sequences of approximately 21 polyomaviruses infecting humans, nonhuman primates, birds, bats, rabbits, and rodents have been deposited in GenBank. Although the roles of this family of viruses in human cancers is still not clear, SV40 and mouse polyomavirus have yielded valuable information about the process of cellular transformation by DNA viruses (Atkin et al, 2009; Gjoerup and Chang, 2010). SV40 and mouse polyomavirus may cause tumors in newborn hamsters but have not been associated directly with human cancer. Mouse polyomavirus was found to cause tumors in the salivary glands of mice and subsequently in many other organs, leading to the name by which it is known. The SV40 virus was identified as a contaminant in poliomyelitis virus vaccines prepared in rhesus monkey cells and caused widespread concern after it was discovered that the virus yielded tumors in newborn hamsters. The virus was injected unwittingly into millions of people. Epidemiological studies of people who received the vaccine gave no evidence that it can cause cancer in humans. However, the fact that SV40 gene sequences were identified in some human tumors, including ependymomas, mesotheliomas, and osteosarcomas renewed fears that the virus might contribute to human carcinogenesis, and the presence of SV40 DNA sequences and T antigens in these tumors has been reported (reviewed in Jasani et al, 2001; Qi et al, 2011). Although it is unlikely that SV40 infection alone causes human malignancy, SV40 may act as a cofactor in the pathogenesis of these tumors. JC and BK viruses are associated with progressive multifocal leukoencephalopathy and interstitial nephritis, respectively, but are less studied. Sporadic reports also describe their presence in some primary brain tumors, osteosarcomas, lymphomas, and colon cancers (reviewed in Boothpur and Brennan, 2010). Most recently, a polyomavirus was shown to be associated with the skin cancer Merkel cell carcinoma using DNA sequencing and digital transcriptome subtraction (Feng et al, 2008). The viral genome is found in cancerous lesions derived from resident Merkel cells of the skin, which comprise the epidermal mechanoreceptors involved in touch discrimination (reviewed in Gjoerup and Chang, 2010; Schrama et al, 2012). It affects immunosup-pressed individuals including the elderly, people with AIDS, and posttransplantation patients, and is normally treated with surgery and radiation. The 5-year survival rate is about 75%, 60%, and 25% for localized, regional, and lymphatic forms of this cancer, respectively.
The polyomaviruses interact with susceptible cells in 2 different ways. In permissive cells that support productive infection, the lytic cycle proceeds in 2 phases: an early phase in which nonstructural, regulatory proteins are synthesized, and a late phase during which viral DNA is replicated, coat protein is made, and progeny virions are assembled. Viral DNA is not integrated in the cellular genome during the lytic cycle. Release of mature virus particles results in lysis and cell death. Monkey cells are permissive for SV40 infection, whereas mouse cells are permissive for mouse polyoma infection. A second type of interaction leads to a small proportion of surviving transformed cells that contain viral DNA integrated randomly into host chromosomes. Transformation occurs more commonly in cells that are unable to efficiently support viral replication. In contrast to normal cells, virus-transformed cells show little or no contact inhibition and therefore grow to high cell density in culture; they give rise to multilayered and disorganized cell colonies, show anchorage-independent growth in a semisolid medium containing agar or methylcellulose, and exhibit a decreased requirement for serum. Cells transformed in culture after infection, give rise to tumors when inoculated into susceptible animals.
Polyomaviruses are small, nonenveloped icosahedral viruses that contain circular, double-stranded DNA genomes of about 5 kbp in length (Fig. 6–1). Early gene products are transcribed immediately after the virus enters the cell, followed by the transport of its genome to the nucleus of the host cell. Late genes are transcribed after viral DNA replication begins, and serve to produce viral structural proteins (VP1, VP2, VP3) and Agno-protein, in the case of SV40. The major capsid protein, VP1 interacts with the host cell receptor, which is believed to be the major histocompatability complex class I protein (MHC I) for SV40, and sialic acid for polyomavirus. Agno-protein is a small multifunctional polypeptide implicated in viral transcription, replication, assembly, and release. It can function as a viroporin, bind to other viral proteins (LT, ST, PP2A), and interacts with cellular proteins (HP1α, FEZ1, Ku70, p53, YB-1). The gene for Agno-proteins is not expressed in all polyomaviruses and its exact mechanism of action remains to be determined (Gerits and Moens, 2012). Viral DNA replication, transcription, and virion assembly occur in the nucleus. The early genes of SV40 include large T antigen, small t antigens, and 17-kDa protein (Fig. 6–1A) and the early genes of polyomavirus are large T, middle T, and small t antigens. These gene products possess transforming properties.
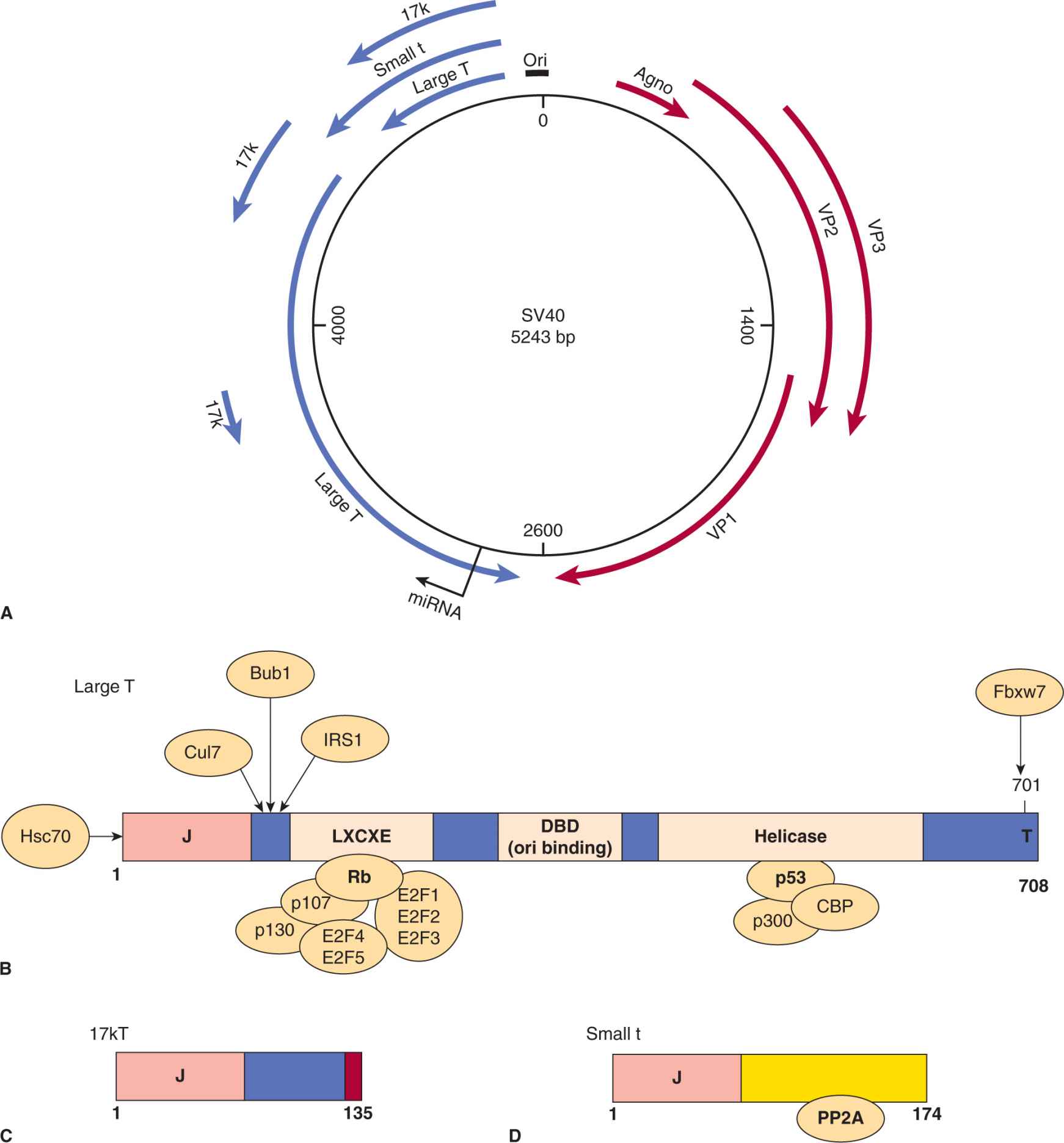
FIGURE 6–1 Simian virus 40 (SV40) gene expression. A) The SV40 genome consists of a circular duplex DNA molecule. Primary transcripts are expressed from opposite DNA strands that constitute early (blue) and late (red) viral mRNA. The largest of the 3 early mRNA spliced transcripts encodes large-T antigen. The shorter early transcripts encode the small t antigen and 17-kDa protein. The aminoterminal 82 residues of the small t and 17-kDa proteins are identical. The late mRNA encoding the VP1, VP2, and VP3 structural proteins and Agno-protein as indicated. B) Schematic representation of the functional domains of large T antigen. Both p53 and pRb, the products of 2 cellular tumor-suppressor genes, interact with large T antigen. The Rb protein binds to the LXCXE motif, which can also bind to the Rb-related proteins p107 and p130. Rb family proteins normally bind and sequester the cellular transcription factors E2F1, E2F2, E2F3, E2F4, and E2F5 inhibiting transcription. Binding of Rb to LXCXE allows the release of E2F factors resulting in transcription and cell-cycle progression into S phase (see Chap. 9, Sec. 9.2.3). The p53 and CBP/p300 proteins bind to a similar region in the helicase domain. T antigen has DNA helicase and adenosine triphosphatase (ATPase) activity for its replication function with cellular DNA polymerase and DNA binding proteins. This protein is the switch between early and late transcription, and T-antigen binds to the origin of replication through the Ori DNA binding domain (DBD). The protein is phosphorylated, binds Zn++, and has a host range or species specific domain (hr/hf) at the extreme carboxyl terminus. C) The 17-kDa protein is a truncated version of large T antigen and contains a J region and RB binding region. It has immortalizing and transforming properties. D) The small t antigen is distinct in sequence and binds and inhibits the cellular phosphatase PP2A.
The large T antigens of the polyomaviruses are complex multifunctional proteins and the functional domains of SV40 T antigen are summarized in Figure 6–1B (reviewed in Cheng et al, 2009; Gjoerup and Chang, 2010). These domains are similar for the closely related large T antigens in all polyoma-viruses. More than 95% of large T antigen is associated with the nucleus but a small portion is found linked to the plasma membrane. The protein is phosphorylated, O-glycosylated, adenosine diphosphate (ADP)-ribosylated, is associated with Zn++, binds adenosine triphosphate (ATP), and possesses a nuclear localization signal (NLS). The large T antigen binds the viral DNA origin of replication in the form of double hexamer structures and functions as the switch between early and late transcription. This protein has DNA helicase activity, unwinds double-stranded DNA, and hydrolyzes ATP in the process. Large T antigen contains 3 domains involved in the transformation and immortalization of rodent cells (Cheng et al, 2009; Gjoerup and Chang, 2010). The N-terminal J domain stimulates both viral replication and enhances oncogenic transformation. It binds specifically to the heat shock protein Hsp70 and activates its adenosine triphosphatase (ATPase) activity during the process of chaperone-mediated protein folding. The J domain also functionally inactivates the p130 and p107 Rb proteins and promotes their degradation. Next to the J domain is another protein region, which contains the LXCXE (leu-amino acid-cysteine-amino acid-glu) motif that is found in proteins that interact with molecules in the Rb family. The Rb protein (p105) and related proteins p107 and p130 function by normally binding to and blocking the functions of E2F transcription factors. Rb preferentially binds to “activating” E2Fs (E2F4/5) whereas p107/p130 bind “repressing” E2Fs (E2F4/5). The large T antigen binds to pRb, p107, or p130, disrupting their regulator roles and activating or repressing specific genes that are associated with cell-cycle progression (reviewed in DeCaprio, 2009). Finally, another transformation domain found in the T antigen of SV40, forms complexes with p53 and led to the discovery of this tumor-suppressor gene. This interaction was originally discovered in 1979 and heralded the recognition of cellular tumor-suppressor proteins (Lane and Crawford, 1979; Linzer and Levine, 1979). T antigen binding to p53 inhibits apoptosis and favors cell-cycle progression. Transfection of the gene encoding large T antigen may alone cause the malignant transformation of normal rodent cells, although the presence of the small t antigen contributes to the full expression of an SV40-transformed phenotype. The T antigen of SV40 can also bind to other factors including cullin 7 (involved in ubiquitination), IRS1 (an insulin-like growth factor receptor), p300/CBP (histone acetylase transcription factors), Bub1 (kinase involved in mitotic checkpoint control), and Fbxw7 (ubiquitination of cyclin E, Myc, Notch, and Jun; see Chap. 8, Fig. 8–9 and Chap. 9, Fig. 9–4). These interactions are summarized in Figure 6–1B. The 3D structures of large T antigen complexed to p53 and the origin DNA was reported (Bochkareva et al, 2006; Lilyestrom et al, 2006). Expression of large T antigen alone, or in combination with small t antigen, will transform most rodent cell types. Transgenic mice that contain the SV40 large T antigen gene develop a high incidence of tumors in organs in which the gene is expressed (reviewed in Saenz Robles and Pipas, 2009).
The small t antigens of SV40 and polyomavirus also function in transformation. Small t antigen is dispensable for lytic growth of the virus but it does favor cell-cycle progression (G1-S) through activation of growth factor signal transduction pathways, via interaction with the catalytic and structural subunits of protein phosphatase 2A (PP2A). By displacing the structural subunit, small t antigen inhibits phosphatase activity and upregulates kinase activity in mitogen-activated protein kinase (MAPK), stress-activated protein kinase (SAPK), protein kinase C (PKC), phophatidylinositol-3 kinase (PI-3 kinase), protein kinase B (PKB/AKT) and nuclear factor kappa B (NF-κB) pathways (Pallas et al, 1990). The middle T antigen of mouse polyomavirus localizes to the plasma membrane and interacts with components of signal transduction pathways including c-src and PI-3 kinase to upregulate MAPK and PKB/AKT activity (Auger et al, 1992; see Chap. 8 for details of pathways). Because middle T antigen also contains most of the sequence of small t antigen, it can also complex with and inhibit the effects of PP2A (Pallas et al, 1990). The interactions of all 3 T antigens with specific cellular components favor cell-cycle progression, cell division, activate growth factor pathways, and inhibit apoptosis. These 3 proteins account for all of the oncogenic properties of the polyomaviruses.
6.2.2 Human Adenoviruses
Adenoviruses are common and can cause acute infections of the upper respiratory tract, infantile gastroenteritis, pharyngitis, and conjunctivitis (reviewed in Berk, 2007). Most people have antibodies directed against 1 or more of these viruses. Aside from adenovirus type 12, which can cause tumors in newborn hamsters, adenoviruses have never been associated with human tumor development. Human cells infected with adenovirus undergo lytic infection, whereas rodent cells are less permissive for growth of the virus and readily survive infection to undergo transformation. The ability of human adenoviruses to induce tumors in rodents and transform cells in culture has rendered them as important tools to study malignant transformation.
Adenoviruses are icosahedral particles with “antennae-like” fibers emanating from the vertices of the icosahedron (Fig. 6–2A) that are composed of 11 structural proteins (Berk, 2007). The 35-kbp linear, double-stranded DNA genome is organized into early and late transcription units (Fig. 6–2B), containing 5 early transcription units (E1A, E1B, E2, E3, and E4), and 1 major late unit that is spliced to generate 5 families of late mRNAs (L1 to L5) that encode the structural viral proteins. Cells transformed with adenoviruses contain an incomplete viral genome that always includes the viral E1A (early region 1A) and E1B genes integrated into host DNA. This minimal region of adenovirus DNA has the capacity to transform rat embryo cells following DNA-mediated gene transfer.
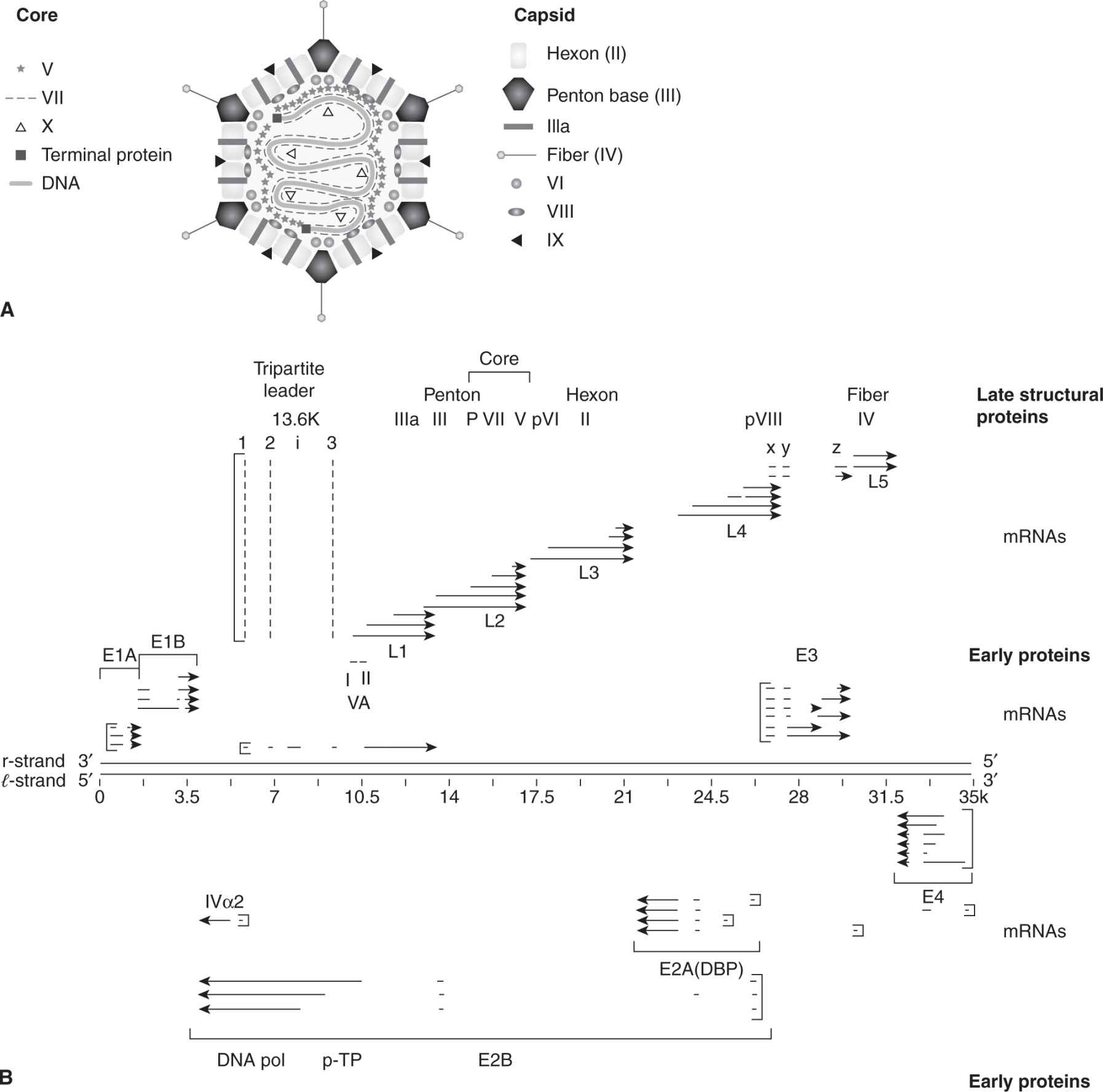
FIGURE 6–2 The structure of human adenovirus and a representation of RNA transcripts and proteins made from the adenovirus genome. A) The virus consists of capsid and core proteins. Core proteins include hexon, penton base, IIIa, fiber, VI, VIII, and IX proteins. The core contains the double-stranded DNA genome, major core proteins (V, VII), terminal proteins, and minor core protein (X). B) The genome is a template for early gene transcripts (E1A, E1B, E2, E3, E4) and late gene transcripts (L1, L2, L3, L4, L5). Early gene products cause transformation, inhibit apoptosis, regulate DNA replication, and block the immune response. Late gene products code for structural proteins (penton, core, hexon, pVIII, and fiber) and mRNA is transcribed from the major late promoter.
Two mRNA species, 12S and 13S, are produced by differential splicing from E1A and encode similar proteins of 243 and 289 amino acids that function as transcription activators. These 2 proteins differ internally by an additional 46 amino acids that are unique to the 13S product. Multiple transcripts also originate from the E1B gene, giving rise to 2 major proteins of 19 kDa and 55 kDa, which can block apoptosis (see Chap. 9). E2 encodes the proteins E2A and E2B that function in DNA replication. E3 proteins are also multiply spliced gene products that are designated by their molecular masses and interfere with immune response. E4 proteins are a diverse family of proteins that mediate transcription, mRNA transport, modulate DNA replication, and inhibit apoptosis.
In gene-transfer experiments, both E1A gene products immortalize primary rodent cells and complement an activated ras gene to transform cells. E1B can replace ras in this type of assay to promote transformation. Additional functions attributed to E1A protein include its ability to either activate or repress transcription from cellular and viral genes dependent on enhancer sequences. Comparison of the E1A amino acid sequence among several adenovirus serotypes shows the presence of 3 conserved regions (CR1, CR2, and CR3). Mutational analysis of the E1A region has revealed that CR1 (amino acids 40 to 80) and CR2 (amino acids 121 to 139) are necessary for transformation, whereas CR3 (amino acids 140 to 188) is dispensable (Moran and Mathews, 1987). The E1A regions required for control of cell growth, blockade of differentiation, and transformation comprise the nonconserved amino terminus together with CR1 and CR2. CR2 contains the LXCXE motif that binds to pRb and is also present in SV40 large T antigen and in the E7 protein of human papillomaviruses. E1A can activate transcription by E2F by promoting its dissociation from pRb (DeCaprio, 2009) and promotes cell-cycle progression, moving the infected cell into S phase. E1A also blocks acetylation of histones and inhibits the transcription of certain genes. Both of these processes are thought to play a role in E1A-mediated transformation.
The E1B55K protein binds p53 and inhibits its transactivation function as well as p53-dependent apoptosis (Harada and Berk, 1999). Unlike E1A, expression of the E1B55K protein alone is insufficient to stimulate resting cells to enter S phase. The E1B19K protein has many activities, all of which tend to block apoptosis and favor cell survival. It is a member of the antiapoptotic Bcl-2 family and has been shown to block both Fas and tumor necrosis factor (TNF)-mediated apoptosis by interacting with Bax (Han et al, 1996). In addition, E1B19K can upregulate SAPK (see Chap. 8, Sec. 8.2.4) to increase transcription and expression of c-jun, promoting cell survival (See and Shih, 1998).
Adenoviruses have been shown to antagonize the immune system. The E1A protein and viral-associated RNAs inhibit the protective effects of interferons α and β. E3 proteins block induction of apoptosis by cytotoxic T lymphocytes (CTLs) and macrophages, and interfere with the processing and presentation of peptide antigens (see Chap. 21, Fig. 21–2). Gene products of the E3 region can be deleted without affecting infections in culture, but these mutations dramatically reduce infections in vivo.
Adenoviruses have been evaluated extensively for cancer gene therapy with multiple reengineering strategies (Pesonen et al, 2011; reviewed in Yamamoto and Curiel, 2010). One of the earliest manipulations was Onyx-015 (or dl-1520), wherein the viruses cannot synthesize the E1B55K protein, facilitating its preferential replication in tumor cells (Bischoff et al, 1996). A similar version of the E1B-55K-deleted adenovirus, called H101, was developed and approved for use in China. It was administered via intratumoral injection in combination with chemotherapy for patients with head and neck squamous cell carcinoma, and demonstrated higher response rates than in patients treated with cisplatin alone (Yu and Fang, 2007). However, because of the limitations of intratumoral injections, these strategies have never evolved into broader applications or lived up to the expectations derived from animal experiments. Furthermore, an unfortunate incident resulted in a fatal outcome in one individual 4 days into a gene therapy trial (Marshall, 1999), which was likely related to a severe immune reaction directed against the adenovirus proteins in the liver. This slowed the development of adenovirus based vectors for gene therapy and oncolytic treatment. In addition the lack of specific viral receptors (CAR) in the tumor, the presence of preexisting antibodies against Ad5 serotype, and the need for more stringent replicative selectivity for cancer cells, have tempered enthusiasm for oncolytic adenovirus therapy (Yamamoto and Curiel, 2010).
6.2.3 Human Papillomaviruses
Human papillomaviruses (HPVs) are nonenveloped DNA viruses that infect epithelial cells to cause warts in the skin, condylomas in mucous membranes, and malignancies of the cervix, vulva, and anal canal, and recently, head and neck cancers (Moody and Laimins, 2010; reviewed in zur Hausen, 2002). Papillomaviruses contain a single molecule of circular double-stranded DNA that is about 8 kb in length, which encodes 8 “early” genes (E1 to E8) and 2 “late” genes (L1 and L2). In addition there is a noncoding regulatory region called the long control region (LCR) (Fig. 6–3A). The functions of the various proteins are as follows: L1 is the major capsid protein whereas L2 is a minor capsid protein that associates with genomic DNA. E1 directs initiation of DNA replication and E2 is a transcription activator that has an auxiliary role in replication. The function of E3 is not known, whereas E4 disrupts cytokeratins and is important for viral release. E5 is a membrane protein with transforming properties that interacts with growth factor receptors, and regulates protein trafficking. E6 is a transforming protein that targets p53 for degradation in the ubiquitin pathway. E7 is a transforming protein that binds to the Rb protein. The function of E8 remains unknown.
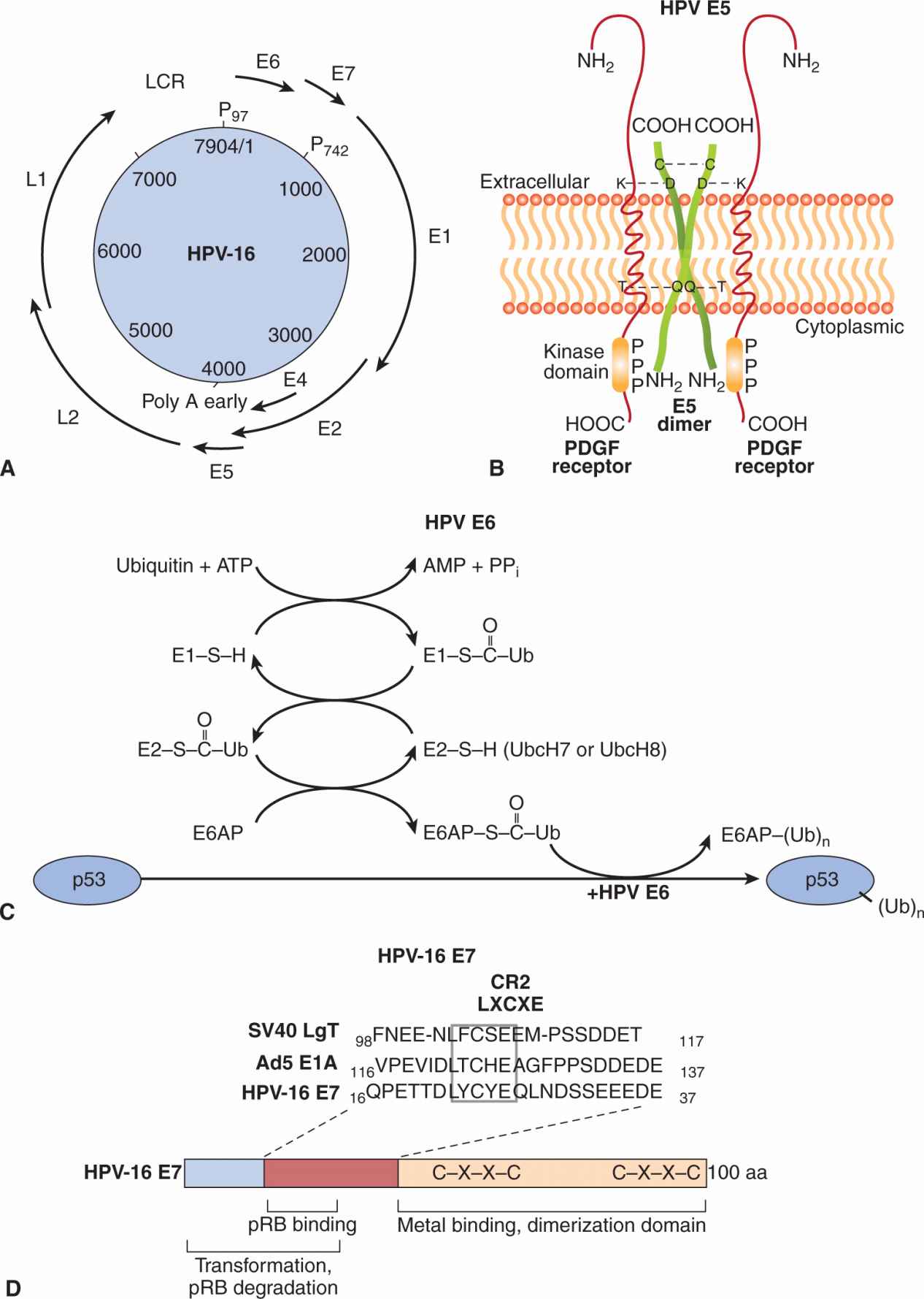
FIGURE 6–3 A) Organization of the HPV-16 genome. The papillomavirus genome is double-stranded circular DNA, but the genes are expressed from only 1 strand in a unidirectional manner. The coding regions for viral proteins in all 3 possible translation phases are indicated by the solid heavy lines and are based on the complete DNA sequence. E and L stand for early and late proteins, respectively. There are 6 open reading frames that code for early proteins (E1, E2, E4, E5, E6, E7) and 2 for the late structural proteins (L1, L2). P97 and P742 are the promoters used in transcription. B) E5 protein of papillomavirus dimerizes through an intermolecular disulfide bond linkage and interacts with platelet-derived growth factor receptor (PDGFR). Aggregation of the receptor occurs through ionic (lysine-aspartic acid, K-D) or hydrogen bond (threonine glutamine, T-Q) interactions to activate the receptor associated tyrosine kinase. C) E6 is a ubiquitin ligase that catalyzes the ubiquitination and subsequent degradation of p53 in the proteosomes. Ubiquitin is then transferred to a ubiquitin conjugating enzyme E2. Ubiquitin is subsequently transferred to E6-associated protein (E6AP), which binds to and ubiquitinates p53 in the presence of HPV E6. Additional ubiquitin molecules can be added to p53 and finally the polyubiquitinated p53 is degraded in the proteasome. D) E7 protein binds pRB through an LXCXE (leu-amino acid-cys-amino acid-glu) motif in a CR2 region along with residues in the CR1 domain. It targets the pRB protein for degradation. An adjacent domain contains metal binding and dimerization motifs that can transactivate some genes recognized by E2F. In many respects, the E7 protein is very similar in structure and function to the SV40 large T antigen and adenovirus E1A protein.
Papillomaviruses have been difficult to study and propagate in culture because they replicate in stratified squamous epithelium, a property that cannot be duplicated in mono-layer cell cultures. The virus reaches the basal layers of the epithelium where it can replicate through a process of mechanical stress and damage to the keratinized epithelium. Virus is taken into the host cell through receptor-mediated endocytosis and is transported to the nucleus. Early gene transcription and translation of early proteins and limited replication of the viral genome occur in the basal squamous epithelial cell. The transcription and production of late capsid proteins (L1, L2), high levels of DNA replication, and virus assembly occur in the keratinized epithelium. Virus is released as the stratum corneum is sloughed from the surface of the skin or mucosa.
More than 100 different types of HPV have been identified. These viruses infect only epithelial cells and are associated mostly with benign mucosal and cutaneous lesions, such as warts in the skin and anogenital regions. Low-risk anogenital warts can be caused by several HPV types—including HPV6, -10, and -11—that infect the genital tract and are associated also with low grades of cervical intraepithelial neoplasia that regress spontaneously; they are rarely found in malignant tumors. Anogenital warts are prevalent among young sexually active adults and their incidence increased 6-fold between 1966 and 1981 during the sexual revolution.
Cervical cancer is the third most common cancer in women worldwide and is a sexually transmitted disease that is associated with high-risk HPV types including HPV16, -18, -31, -33, and -45, that are found in approximately 90% of all cervical cancers (reviewed in zur Hausen, 2002). However, these HPV types have also been detected in nonmalignant cervical tissue, and only a small proportion of women with clinically apparent high-risk HPV infection eventually develop cervical carcinoma, possibly because many of these infections may be transient. HPV infection is not sufficient for tumor development and other cofactors—such as smoking, use of oral contraceptives, recurrent infection, early pregnancy, and immunological and hormonal status—may play a role in progression to malignancy. In addition to the aforementioned malignancies, HPV has also been associated with nonmelanoma skin cancers, penile cancer, and respiratory malignant papillomas. Immunosuppressed individuals, transplant recipients, and AIDS patients are at higher risk for HPV-related malignancies.
There has been increasing recognition of HPV involvement in head and neck cancers, particularly those located in the oropharyngeal region such as the tonsils, and the base of tongue (Chung and Gillison, 2009). Approximately 60% to 70% of oropharyngeal squamous cell carcinomas diagnosed in North America are associated with HPV. There are multiple purported reasons for this increasing incidence, such as reduction in smoking, but other lifestyle issues likely contribute to this epidemic (D’Souza et al, 2007). Dissimilar to cervix cancer, greater than 90% of HPV involvement in oropharyngeal cancers is dominated by HPV16. Furthermore, HPV-associated oropharyngeal cancers overexpress the cyclin-dependent kinase (CDK) inhibitor CDKN2A or p16, such that p16 immunohistochemistry on formalin-fixed patient biopsies is a diagnostic surrogate for identifying such HPV-infected entities (Shi et al, 2009). HPV-associated oropharyngeal cancers have a superior clinical outcome, with 3-year overall survival rates of approximately 82% to 85% versus 60% to 65% between HPV-positive and HPV-negative cancers (Shi et al, 2009; Ang et al, 2010). Many possibilities have been suggested to explain this difference, including better performance status of patients (younger, more likely to be nonsmokers); increased expression of p16 hence lower proliferative potential; less deleterious p53 mutations (Westra et al, 2008), and HPV immunological responses. Whole exome sequencing studies have documented that HPV-positive tumors harbor half the mutation rates of HPV-negative malignancies (Stransky et al, 2011). The specific mechanisms remain to be elucidated, but explain the better treatment outcomes for patients with HPV-positive tumors.
The HPV genome is maintained in benign warts as an episome (a nonintegrated, circular form). In malignant cells, HPV DNA is integrated randomly into various chromosomes, resulting in substantial deletions or disruption of the viral genome, particularly the E2 gene, which has a negative regulatory effect on the expression of the HPV proteins E6 and E7. These latter 2 proteins are always retained and consistently expressed in cervical tumor tissue and cell lines, suggesting that 1 or both of these proteins may be required for transformation by HPV. A comparison of high-risk HPVs, such as HPV16 and HPV18, with low-risk viruses, such as HPV2, HPV4, HPV6, and HPV11, has allowed the mapping of transformation properties to E5, E6, and E7 genes. The E5 protein can dimerize and interact with growth factor receptors, such as those for the epidermal growth factor receptor (EGFR) and platelet-derived growth factor receptor (PDGFR), leading to activation of the MAPK pathway (see Chap. 8, Sec. 8.2.4) and cell proliferation (see Fig. 6–3B). Gene transfer experiments with primary human fibroblasts or keratinocytes demonstrate that either the E6 or E7 genes from the high-risk HPV types, but not from the low-risk HPV types, extend the life span of these cells in culture (Hawley-Nelson et al, 1989; Munger et al, 1989). These cells, however, will not form tumors when injected into nude mice and enter a crisis period in culture. HPV E6 proteins interact with and inactivate the p53 tumor-suppressor protein (see Fig. 6–3C). E6 may have additional functions that operate in the transformation process as it can promote hyperplasia in a p53 null background. HPV E7 proteins contain domains similar to the adenovirus E1A protein and large T antigen of SV40; the transforming ability of E7 proteins depends on their binding to pRb tumor-suppressor proteins (Dyson et al, 1989) through a domain that contains the LXCXE motif (see Fig. 6–3D). Binding of E7 to pRb favors the degradation of the tumor suppressor and results in the release of the bound E2F transcription factor from pRb to induce cellular DNA synthesis and progression into the S phase of the cell cycle. The E7 protein from low-risk HPV types bind to pRb with at least 10-fold lower efficiency than the E7 protein of HPV16 and HPV17. The E7 protein can also interact with CDK inhibitors such as p27 and p21 (see Chap. 9, Sec. 9.2.2), which may promote the replication of papillomavirus DNA in differentiating squamous epithelial cells. Thus, related mechanisms of transformation seem to be evident for papovaviruses, adenoviruses, and papillomaviruses (see Fig. 6–3D).
Prophylactic vaccines directed against the high-risk papillomaviruses have been evaluated and marketed. Gardasil (Merck Pharmaceuticals) is a recombinant quadrivalent vaccine consisting of virus-like particles prepared from synthetic L1 proteins and protects against HPV6, -11, -16, and -18 after a regimen of 3 injections. This vaccine has been demonstrated to be effective for both young women and men at reducing the incidence of intraepithelia neoplastic and external genital lesions, respectively (Future II Study Group, 2007; Giuliano et al, 2011). A similar vaccine, Cervarix (GlaxoSmithKline), designed to target HPV types 16 and 18, is similarly efficacious (Paavonen et al, 2009), is also able to cross-protect against HPV31, -33, -45, and -51 (Wheeler et al, 2012). Despite success in development of these HPV vaccines, and the efficacy of primary prevention, uptake of these vaccines in developed countries ranges from 50% to 80%, indicating that there remain important challenges in these diseases (Ogilvie et al, 2010).
6.2.4 Epstein-Barr Virus
An aggressive lymphoma that affects African children was first described by Dennis Burkitt and is known as Burkitt lymphoma. Cultured cells from these lymphomas were found by Epstein and Barr to release a herpes virus that subsequently became known as Epstein-Barr virus (EBV). EBV is transmitted horizontally, usually via contaminated saliva, infecting more than 90% of the human population by the age of 20 years, often without any manifestation of disease (reviewed in Kieff and Rickinson, 2007; Rickinson and Kieff, 2007). EBV strains can be classified into two main types, type 1 and type 2, based on polymorphisms (genetic differences) within certain viral genes. Type 1 strains are more common in western countries, whereas both type 1 and type 2 strains are prevalent in central Africa and New Guinea. Strong epidemiological and clinical data have associated EBV infection with 3 lymphoproliferative diseases of B-cell origin—infectious mononucleosis, Burkitt lymphoma, and lymphoma of the immunocompromised host, particularly in the setting of organ transplantation and HIV infection. There is also a very strong association between EBV infection and undifferentiated nasopharyngeal carcinoma (NPC), and evidence has implicated EBV in the pathogenesis of Hodgkin disease, T-cell lymphoma, and some gastric carcinomas. Recent epidemiological studies have also implicated the virus in multiple sclerosis (Owens and Bennett, 2012). Geographic and ethnic variation has been recognized in the incidence of EBV-associated malignancies, indicating involvement of other genetic and environmental factors.
EBV contains a double-stranded DNA genome that codes for immediate-early, early, and late gene products. The B lymphocyte is the preferential target cell of EBV and the complement regulatory protein CD21, which normally protects a cell from self-destruction, and MHC II, involved in antigen presentation, are the cell receptors for this virus. CD21 binds to the viral glycoprotein gp350/220, whereas MHC II binds gp42. Two forms of infection can occur in the B cell using 2 different sets of gene products and promoters: (a) latent infection, which is accompanied by B cell proliferation, and (b) lytic infections, which are characterized by synthesis of structural proteins, generation of linear viral genomes, packaging, and the generation of mature virions.
Upon entry into the host cell, the viral genome travels to the nucleus and circularizes to establish a latent infection (Fig. 6–4A). Latency is characterized by the synthesis of 6 EBV nuclear antigens (EBNA-LP, EBNA-1, EBNA-2, EBNA-3A, EBNA-3B, and EBNA-3C). Three latent membrane proteins (LMP1, LMP2A, and LMP2B) are also synthesized during latency. EBNA-1 protein is required for DNA replication of the extrachromosomal viral plasmids in EBV-infected cells. It binds to the viral origin of replication and is essential for maintenance of multiple viral genomes in an episomal form. EBNA-1 also binds to chromosomes to enable the EBV episome to partition to progeny cells. EBNA-LP and EBNA-2 activate transcription from viral and cellular genes (including c-myc; see Chap. 7, Sec. 7.5.2). EBNA-2 protein transactivates expression of cellular and viral genes through interaction with at least 2 sequence-specific DNA-binding proteins. LMP1 has transforming effects in rodent fibroblast cell lines; it permits them to grow under low serum conditions, generate colonies in soft agar, and allows them to form tumors in nude mice (Li and Chang, 2003). In B-lymphocytes, it causes cell clumping, increased villous projections, upregulates vimentin and other proteins, and protects cells from apoptosis through induction of Bcl-2 (see Chap. 9, Sec. 9.4.2). LMP-1 can also cause hyperplasia and lymphomas when expressed as a transgene in mice. LMP1 spans the membrane 6 times and mimics the function of TNF receptor family member, CD40. It acts as a constitutively active receptor that activates NF-κB, p38/MAPK, Jun kinase (JNK), JAK3, and PI3K (see Chap. 8, Secs. 8.2.4 and 8.2.5). The long cytoplasmic tail of LMP1 contains 3 C-terminal activating regions (CTAR1, CTAR2, and CTAR3) which interact with tumor necrosis factor receptor-associated factors (TRAFs) 1, 2, 3, and 5, TRAD1, RIP, and JAK3. Upregulation of the protein kinase pathways can stimulate the production of vascular endothelial growth factor (VEGF), interleukin (IL)-6, IL-8, CD40, fibroblast growth factor (FGF), EGFR, phosphorylation of p53, activation of Akt/PKB, and expression of DNA methyltransferase and telomerase (Soni et al, 2007; Kung et al, 2011). LMP2 proteins span the membrane of the host cell 12 times and mimic the function of the B-cell receptor (BCR; see Chap. 21, Sec. 21.3.1). LMP2A protein possesses a 119-amino acid cytoplasmic N-terminal domain that acts as a substrate for and associates with src family tyrosine kinases (Syk, Lyn). It is required to establish and maintain EBV latency and its expression in nude mice can transform epithelial cells and produce tumors. The function of LMP2A is to block activation of lytic EBV infection by sequestering the protein kinases that would mediate reactivation following crosslinking of the surface immunoglobulin that serves as the BCR (Longnecker, 2000; Dykstra et al, 2001). This association targets the kinases for ubiquitin mediated degradation. LMP2A can also activate PI3 kinase and PKB/AKT (see Chap. 8, Sec. 8.2.5) to enhance survival of the latently infected B cell. LMP2B lacks the amino terminus, involved in the interaction with protein kinases, and interacts with LMP2A to dampen its effect. The LMP2B protein is important in reactivation of the lytic phase of EBV infections.
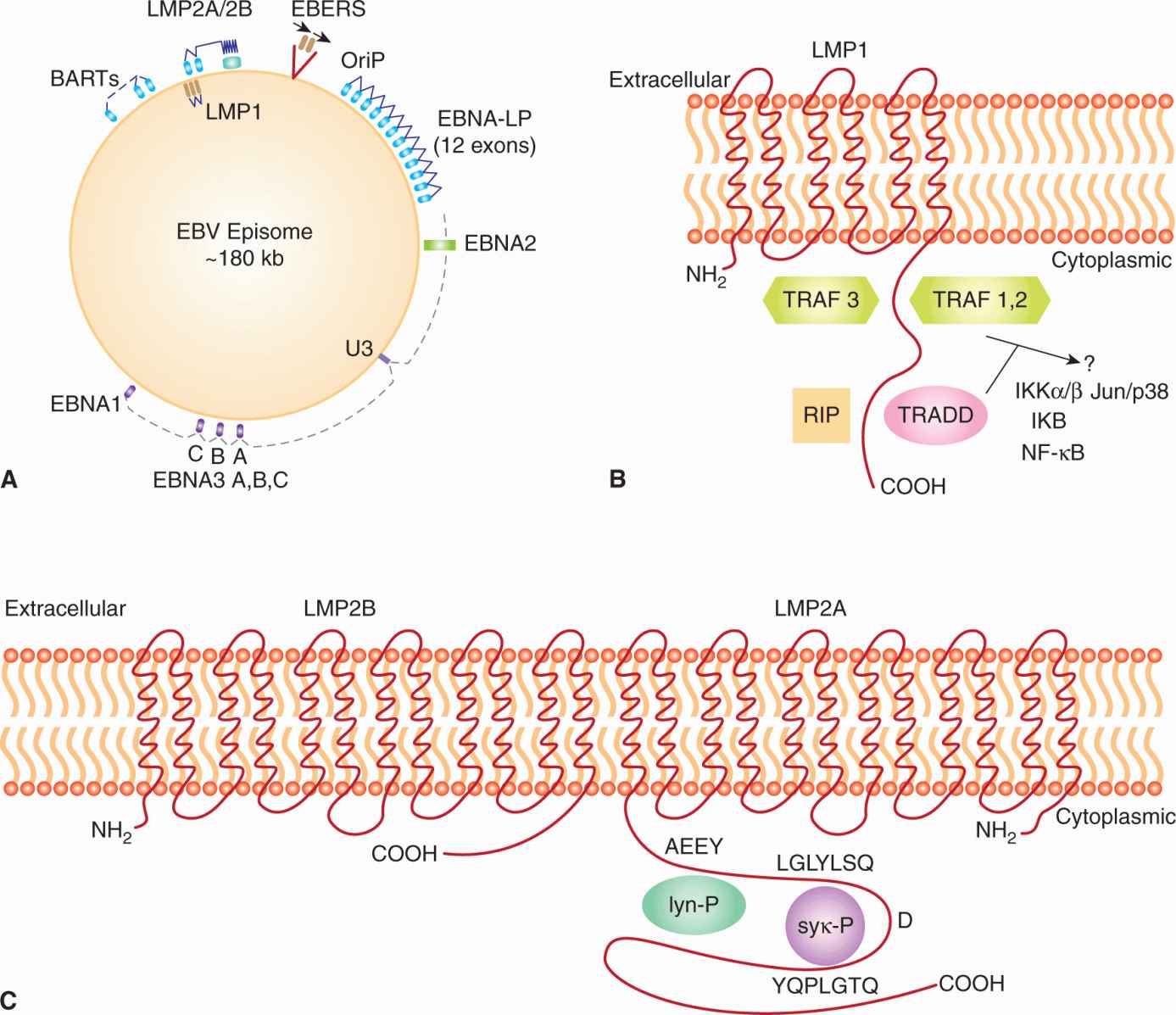
FIGURE 6–4 A) Latency-associated genes of the genome from EBV. When EBV infects a cell, the genome becomes circularized and forms an episome or plasmid. The gene products associated with latency are called Epstein-Barr nuclear antigen (EBNA); leader protein 1, 2, 3A, 3B, 3C; latent membrane protein (LMP) 1, 2A, 2B; Epstein-Barr early RNAs (EBERS); and BamHI RNA transcripts (BARTs). The transcribed gene products are multiply spliced and the exons are represented as gray rectangular boxes. The latency-associated origin of replication is OriP. B) LMP1 is a transforming protein that activates transcription and stimulates cell growth through interaction with TNF receptor-associated proteins (TRAF, TRADD, RIP). It stimulates cell survival pathways by activating NF-κB through phosphorylation of inhibitor of NF-κBNF-κB (IKB) and activation of c-jun. C) LMP2A is receptor-like protein that contains tyrosine phosphorylation domains that sequester src-like tyrosine kinases and prevent reactivation of the infected B cell from its latent state.
Small RNAs are also produced by EBV during infection. The small nonpolyadenylated noncoding RNAs (Epstein-Barr early RNAs [EBERs]) are abundant viral transcripts that bind to double-stranded RNA-activated protein kinase (PKR) and inhibit its activity during the interferon-mediated antiviral response (Iwakiri and Takada, 2010). They also help block apoptosis through the induction of BCL-2 (see Chap. 9, Sec. 9.4.2). Viral micro-RNAs (miRNAs) were first discovered in EBV (Pfeffer et al, 2004; see Chap. 2, Sec. 2.4.3). They have since been found in other herpes viruses including Kaposi sarcoma herpesvirus (KSHV) and cytomegalovirus. There are more than 45 potential EBV miRNAs encoded by either the BamH1 fragment rightward open reading frame (BHRF) or BamHI RNA transcript (BART) regions of the genome. Interestingly, the BART miRNAs are predominantly expressed in cells derived from NPCs, whereas the BHRF miRNAs are expressed in B lymphocytes. These miRNAs appear to regulate viral replication, control transition between latent and lytic infections, block host innate/adaptive immunity, modulate growth of the host cell, or inhibit apoptosis (Cai et al, 2006; Grundhoff et al, 2006; Xing and Kief, 2007). Targets for the miRNAs include viral DNA polymerase, LMP-1, chemokines (CXCL-11), NK cell ligand (MICB), and a p53-dependent modulator of apoptosis (PUMA).
Viral latency is designed to promote the survival of the EBV episome in the face of an efficient attack by cytotoxic T-lymphocytes of the infected cell. Latency attempts to minimize the number of viral proteins and antigenic epitopes being expressed and yet retain the genetic material of EBV within the infected cell (see Chap. 21, Sec. 21.3). Initially the circularized viral genome expresses all the proteins and RNA transcripts associated with latency. This promotes a state of lymphoproliferation and replication of the viral episome. This state, referred to as latency III, is present in the rapidly proliferating B lymphocytes found in infectious mononucleosis or posttransfusion lymphoma. In latency states I and II, fewer viral proteins are expressed (they include EBNA1 and EBERs); this is the characteristic state of NPC cells, T-cell lymphomas, and gastric carcinomas. In vitro this state can be mimicked by fusing EBV-infected lymphoblastic cell lines with EBV-negative epithelial, fibroblast, or hematopoietic cells. Latency II alone is characteristic of cells in Hodgkin disease.
Reactivation of EBV from the latent state to produce structural proteins and virions can occur through a number of stimuli. These stimuli include crosslinking of BCRs with antibodies or treatment with phorbol esters, which mimic phosphatidyl inositol/diglyceride, and activate MAPK and PKC, leading to transcription of a number of genes that are involved in DNA replication. Late genes are subsequently transcribed and specify the structural proteins of EBV. In addition, an IL-10 analog (BCRF1), is transcribed in the late phase of infections and has B-cell growth properties, downregulates natural killer (NK) cells, blocks cytotoxic T-cell activity, and inhibits interferon-γ production to favor a T-helper (Th)-2 immune response by interfering with CD4+ T and NK cell functions (Hsu et al, 1990; Jochum et al, 2012; see Chap. 21, Sec. 21.3.4). Cooperation between the latent and lytic states of EBV infection is an efficient mechanism by which to propagate the viral genome and minimize the exposure of viral proteins to immune surveillance. The proteins made during latency are associated with a variety of diseases, including mononucleosis, Burkitt lymphoma, NPC, Hodgkin disease, and gastric carcinoma.
Infectious mononucleosis is a benign disease where symptoms range from mild transient fever to several weeks of pharyngitis, lymphadenopathy, swollen spleen and lymph nodes, and general malaise. Infectious mononucleosis is characterized by the proliferation of latently infected B cells coupled with the effects of reactive T cells, which eventually clear the pathogen. EBV is released from the oropharynx in localized lytic infections that appear to occur when latently infected B cells are in contact with mucosal surfaces. Linear EBV DNA, characteristic of lytic infection, is detectable in only a fraction of tonsillar lymphocytes. Epithelial cells in these local regions may become permissive to EBV infection. At the peak of an acute infection, 0.1% to 1% of the circulating B cells are positive for EBERs and the latency antigens. Expanding B-cell populations carry somatically rearranged immunoglobulin (Ig) genes that are characteristic of antigen primed polyclonal memory B cells and are capable of producing antibodies (see Chap. 21, Sec. 21.3.1). Upon resolution of infectious mononucleosis through the action of cytotoxic T-lymphocytes directed against both lytic and latent antigen epitopes, an asymptomatic carrier state is established.
Burkitt lymphoma is the most common childhood cancer in equatorial Africa and is also found in coastal New Guinea (Gromminger et al, 2012; reviewed in Molyneux et al, 2012). Climatic factors, the high incidence of Plasmodium falciparum malaria, as well as EBV infection, predispose children in these areas to the disease. Both type 1 and type 2 isolates of EBV have been found to be associated with Burkitt lymphoma. Tumors present at extranodal sites, most frequently in the jaw during molar tooth development, but also in the orbit of the eye, the central nervous system, and in the abdomen. It affects 3 times as many boys as girls. The tumors are monoclonal, are composed of germinal center B cells, and contain chromosome translocations between c-myc and the IgG heavy-chain locus (8:14) or c-myc and the IgG light-chain loci (8:2 or 8:22). These translocations are believed to result in deregulation of c-myc expression as a result of proximity to the immunoglobulin enhancer sequences, thus preventing the normal downregulation of c-myc expression in maturing B lymphocytes. Malaria or AIDS together with EBV may supply lymphoproliferative signals that favor translocations. High c-myc expression in the EBV-transformed B cell can substitute for the growth-promoting effects of the EBV latency genes. Burkitt lymphoma is endemic in Africa, but it can also occur, although 50 to 100 times less frequently, in Europe and North America. A third form of the disease now affects patients infected with HIV; it is called AIDS-related Burkitt lymphoma. The lymphomas are “immunologically silent” and avoid surveillance by cytotoxic T lymphocytes through inhibition of proteosome-dependent antigen presentation, downregulation of MHC I, and low levels of costimulatory and cell adhesion molecules on the cell surface (see Chap. 21, Secs. 21.3.5 and 21.3.6). Normally only EBNA1 and EBERs are expressed in Burkitt lymphoma, accounting for a lack of immunogenicity against the tumors. In 30% of tumors, the p53 gene is also mutated.
Nasopharyngeal carcinoma is a relatively rare disease in Europe and North America but is almost endemic among Southeast Asian populations, particularly those of southern China. The link between NPC and EBV was first suggested by the presence of elevated antiviral antibodies in the sera of patients. DNA hybridization studies and polymerase chain reaction (PCR) amplification have subsequently confirmed that EBV is present in most tumor samples. Real-time quantitative PCR can be used to measure EBV DNA load in the plasma, and can be correlated with tumor burden and recurrence following treatment (Lin et al, 2004). The role of EBV in NPC is continuing to be elucidated; racial, genetic, and environmental cofactors all appear to be important (Lo et al, 2004). MHC I haplotype, viral LMP1 variants, dietary factors (nitrosamines in dried fish), chemical carcinogens, cytochrome P450 mutations, and physical irritants (dust and smoke) are all implicated as risk factors for NPC. Four EBV proteins, in addition to EBERs, have been detected in NPC cells, namely the nuclear antigen EBNA-1, LMP1, LMP2A, and LMP2B. The profound growth-stimulating effect of LMP1 on keratinocyte cultures suggests that LMP1 expression may exert similar effects in the nasopharyngeal epithelium. The miRNAs derived from the BART transcripts are very abundant in these tumors and probably play a significant role in carcinogenesis (Marquitz and Raab-Traub, 2012). With advances in both diagnostic and therapeutic technologies such as intensity-modulated radiation therapy (IMRT), the clinical outcome for NPC has improved over the past decade, especially for locoregional control, although challenges remain in addressing the development of distant metastases (Razak et al, 2010).
Hodgkin disease is a lymphoma characterized by a malignant population of mononuclear B and multinuclear Reed-Sternberg cells set within a background of reactive nonmalignant lymphocytes. Reed-Sternberg cells carry the genotype of a cell that has inappropriately escaped apoptotic death and have the properties of monoclonal postgerminal center (memory) B cells. They do not express conventional B- or T-cell markers. Reed-Sternberg cells express a range of cytokines that have the capacity to divert CTL recognition away from them. An association between EBV and a subset of patients with Hodgkin disease is now supported by circumstantial evidence. EBV DNA sequences and transcripts have been detected in malignant Reed-Sternberg cells and their mononuclear variants by in situ hybridization and PCR-based assays (reviewed in Kapatai and Murray, 2007). LMP1 protein has also been detected by immunohistochemical staining of lymph nodes from patients with Hodgkin disease. The association of EBV with this disease varies greatly from country to country, and Hodgkin disease in developing countries differs from that in Western countries in terms of epidemio-logical, pathological, and clinical characteristics. Thus, 100% of Kenyan children with Hodgkin disease were found to be EBV-positive (53 of 53 cases), whereas 51% of children from the United States and the United Kingdom (46 of 90 cases) showed evidence of EBV in the malignant cells.
Gastric carcinoma is caused by EBV in approximately 10% of all cases, and is associated with the lymphoepithelial subtype of stomach cancer (reviewed in Fukayama, 2010; Jang and Kim, 2011). It is characterized by the monoclonal growth of EBV-infected epithelial cells that express EBNA1, EBERs, BART transcripts, and small amounts of LMP2A, which is characteristic of latency state I. The LMP2A protein is believed to cause methylation of CpG motifs in the promoter region of the PTEN tumor suppressor (see Chap. 7, Sec. 7.6.2). More explicitly, LMP2A induces the phosphorylation of STAT3, which activates transcription of DNMT1 methyltransferase, and causes loss of PTEN expression through CpG island methylation of the PTEN promoter in EBV-associated gastric carcinoma. Virus-derived miRNAs are also believed to contribute to gastric carcinoma. EBV-associated gastric carcinoma affects 75,000 patients worldwide each year.
6.2.5 Hepatitis B Virus
Most individuals infected with the hepatitis B virus (HBV) develop either an acute transient illness or an asymptomatic infection that leaves them immune. However, severe liver failure associated with fulminant hepatitis can occur in approximately 1% of those infected with HBV and usually results in death. Approximately 10% of infected individuals develop chronic hepatitis, which can progress to more severe conditions, such as cirrhosis and liver cancer. There are estimated to be 500 million chronic carriers of HBV worldwide and 2 billion seropositive individuals have been exposed to the virus. There is strong epidemiological evidence indicating the importance of chronic HBV infection in the development of human hepatocellular carcinoma. Liver cancer is the fifth most common malignancy worldwide and the third leading cause of cancer death. More than 80% of individuals with liver cancer have been chronically infected by HBV. Hepatitis B is widespread throughout Asia, Africa, and regions of South America, and in some regions hepatocellular carcinoma is the leading cause of cancer death. Transmission of HBV is primarily through blood and sexual contact. In Asia, vertical transmission from mother to child during the birthing process is the main route of infection. Introduction of the recombinant vaccine derived from the membrane protein of HBV (Recombivax), which is administered as a 3-dose regimen, has limited the incidence of this virus in North America and dramatically reduced hepatitis B in the younger populations throughout Asia (Cassidy et al, 2011; Luo et al, 2012; Xiao et al, 2012).
HBV is an enveloped DNA-containing virus (reviewed in Seeger et al, 2007). The envelope contains 3 related forms of surface-exposed glycoproteins (L, M, S) that act as the major surface antigenic determinant (hepatitis B surface antigen [HBsAg]; Fig. 6–5A). The larger surface antigen (L) interacts with an unidentified HBV receptor on the plasma membranes of hepatocytes. Viral membranes surround an icosahedral nucleocapsid composed of core protein (C). This nucleocapsid contains at least 1 HBV polymerase protein (P) as well as the HBV genome. Viral DNA found in viral particles is composed of one 3.2-kb strand (minus strand) base-paired with a shorter “plus” strand of variable length. Small fragments of remnant RNA pre-genome can also be found in the virus as a consequence of replication. Because the 5′ ends of both strands invariably overlap by about 300 bases, the DNA retains a circular configuration, although neither strand is itself a closed circle. The genome of HBV is organized very efficiently and its coding capacity consists of 4 overlapping reading frames that specify core protein (C), envelope surface proteins (L, M, S), polymerase (P), and the viral oncogene (X) (Fig. 6–5B). Different HBV proteins are generated by the transcription and translation of mRNAs from several start sites and in-frame initiation codons.
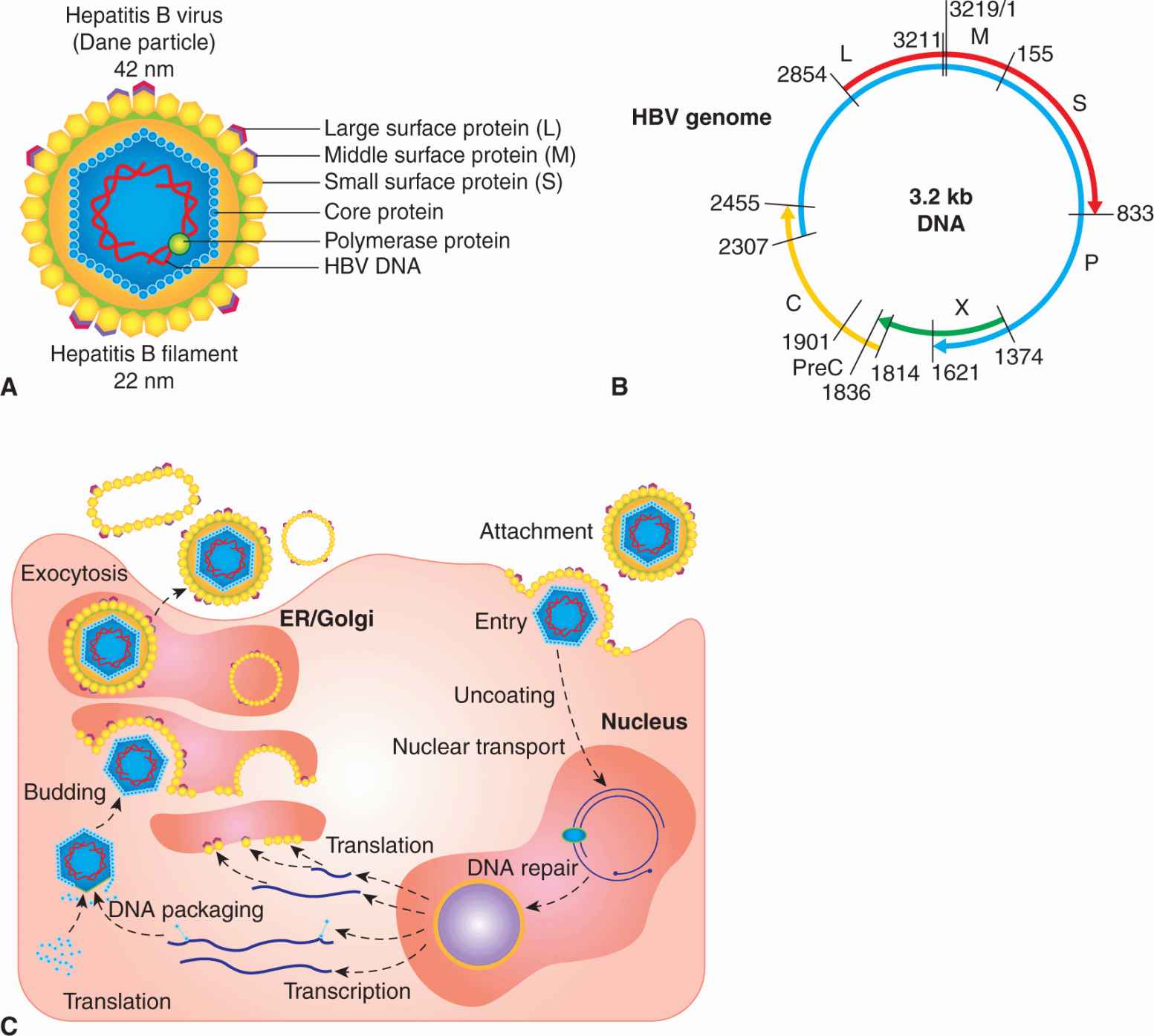
FIGURE 6–5 A) Essential features of the structure of hepatitis B virus. The envelope contains surface-exposed glycoproteins (L, M, S), and surrounds an icosahedral nucleocapsid composed of core protein. The nucleocapsid contains at least 1 HBV polymerase protein, as well as the HBV genome. Viral DNA in the virion is composed of one 3.2-kb strand (minus strand) base-paired with a shorter “plus” strand of variable length. B) The genome of HBV is compact and consists of 4 overlapping reading frames that specify core protein (C), envelope surface proteins (L, M, S), polymerase (P), and the viral oncogene (X or HBx). Different HBV proteins are generated by transcription and translation of mRNAs from several promoter start sites and in-frame initiation codons. A larger form of core protein (precore [PreC]) possesses a signal peptide, causes it to associate with the endoplasmic reticulum (ER), and allows the core antigen (hepatitis B e antigen [HBeAg]) to be secreted from the cell. The shorter form of core protein remains in the infected cell and functions as the major nucleocapsid protein. The X protein is a multifunctional polypeptide that acts as a weak oncogene in HBV infections. C) HBV initiates infection by binding to an unknown receptor on the hepatocyte and the membranes of the virus fuses with that of the host cell. The nucleocapsid core enters the cytoplasm and is transported to the nucleus where the viral DNA (single-stranded DNA gap) is repaired by host enzymes to form a double-stranded circle. Promoters within the HBV DNA are recognized by host cell RNA polymerase to produce RNA transcripts required for viral protein synthesis (C, S, P, X). Host RNA polymerase also directs the synthesis of an RNA pregenome that also serves as a template for the viral DNA genome. PreC is a signal sequence that allows HBeAg to be secreted.
HBV cannot be propagated in cultured cells, and many of the steps of viral replication, such as attachment, host cell entry, and virion assembly, are poorly understood. However, several cell lines are capable of supporting HBV DNA synthesis, following transfection with viral DNA. Genome replication is complex and occurs after viral DNA is transported into the nucleus where the gap is repaired to produce covalently closed-circular DNA by using host cell enzymes (see Fig. 6–5C). Integration of HBV DNA into host chromosomes is not required for replication. Once recircularized, enhancers and promoters within the HBV DNA direct the synthesis of RNA transcripts required for viral protein synthesis (C, S, P, X) and the formation of an RNA pregenome that serves as a template for viral DNA replication. The viral polymerase (P) is actually a reverse transcriptase that first directs the synthesis of minus strand DNA from the RNA pregenome. A ribonuclease (RNase) activity associated with the polymerase subsequently degrades the RNA pregenome, and the viral polymerase uses the RNA fragments to prime synthesis of the positive-sense DNA strand. Completion of this positive strand DNA never occurs and the virus is secreted from the host cell by a process of exocytosis following virus assembly.
HBV likely causes cancer by a combination of virus-specific and host-related factors and 90% of hepatocellular carcinoma (HCC) cases develop in a cirrhotic liver (reviewed in Tsai and Chung, 2010). After 20 to 30 years of chronic infection, 20% to 30% of patients develop liver cirrhosis and liver cancer develops at an annual rate of 3% to 8% in these individuals. During the process of liver injury and inflammation, hepatic stellate cells become activated and transform into myofibroblast-like cells and produce liver fibrosis. Hepatocarcinogenesis is a complex multistep process involving genetic and epigenetic alteration, activation of cellular oncogenes, inactivation of tumor suppressors, and dysregulation of multiple signal transduction pathways. These include Wnt/β-catenin, p53, pRb, Ras, MAPK, Janus kinase (JAK), STAT, PI3K/Akt, Hedgehog, epidermal growth factor (EGF), transforming growth factor (TGF)-β pathways (see Chap. 8). Viral gene products (HBx, preS2/S), random insertion of the viral genome into host chromosomal DNA, liver injury and inflammation, alcohol, and dietary carcinogens together constitute a multifactorial cause for liver cancer.
Random integration of HBV DNA fragments occurs in more than 85% of HBV-related HCC and precedes tumor formation. This process contributes to chromosomal deletions, translocation, produces fusion transcripts, causes DNA amplification, and results in host genomic instability. Insertion of virus DNA can activate human cyclin A2, retinoic acid receptor β, telomerase reverse transcriptase (TERT), platelet-derived growth factor (PDGF) receptor, signaling, and 60S ribosomal protein gene expression. Most integrated HBV DNA fragments contain the HBx and preS2/S genes, which can both contribute to hepatocarcino-genesis. Both these genes can upregulate signal transduction pathways and stimulate transcription of genes characteristic of growth and survival.
HBx is a multifunctional protein whose exact role in HBV infections and carcinogenesis has remained elusive (reviewed in Wei et al, 2010; Kew, 2011; Ng and Lee, 2011). It was originally called the “promiscuous transactivator” because HBx increases the synthesis of many viral and cellular gene products. The X protein is conserved in all mammalian, but not avian, hepadnaviruses, and HCC is only associated with the mammalian viruses. HBx compromises nucleotide excision repair by binding to and inactivating the UV-damaged DNA-binding protein (DDB1), which is also a component of several E3 ubiquitin ligase complexes (see Chap. 8, Fig. 8–9 and Chap. 9, Fig. 9–4). HBx could also help target the E3 ubiquitin ligase to critical cell proteins for ubiquitination and degradation. This interaction could affect STAT proteins and innate immunity, cell-cycle progression, chromosome segregation and remodeling, and produce further mutations that cause HCC. Thus, the interaction of HBx with DDB1 could impinge a number of cellular processes that could lead to HCC (Li et al, 2010; Martin-Lluesma et al, 2008). HBx affects cell signaling pathways including PKC, JAK/STAT, PI3K, SAPK, JNK, MAPK, AP-1, NF-κB, Smad, Wnt, PI3K/Akt, p53, and TGF-β (see Chap. 8). HBx can increase cytosolic calcium levels and activate calcium dependent proline rich tyrosine kinase 2, which, in turn, activates Src kinase (Yang and Bouchard, 2012). HBx decreases proteasomal degradation of β-catenin and interacts with Pin1, a Wnt signal regulator. HBx can cause transcriptional repression of the p53 gene and bind to p53 protein to inhibit its activity. TGF-β expression is upregulated by HBx in HCC tissue and shifts signaling to the oncogenic Smad3L pathway. Hepatic steatosis is also induced by HBx through transcriptional activation of the serum response factor SREBP1. HBx is a very weak oncogene, and can only lead to increased hyperplasia, malignant transformation of hepatocytes, and liver carcinoma in some strains of HBx transgenic mice. Similarly, transgenic mice containing the entire HBV genome do not spontaneously develop HCC. The only satisfactory in vivo experimental system for studying HCC caused by a hepadnavirus is the woodchuck hepatitis virus model.
HBV and hepatitis C virus (HCV, see Sec. 6.3.6) account for 90% of the approximately 6 million cases of HCC worldwide. Globally, it is the fifth most common cancer and the third leading cause of cancer death. Surgical resection and liver transplantation are the most effective treatment options, but the prognosis is usually bleak.
6.2.6 Kaposi Sarcoma-Associated Herpesvirus
KSHV, or human herpesvirus 8 (HHV-8), was identified as the pathogen responsible for a number of malignancies, including Kaposi sarcoma (KS), body cavity-based or primary effusion lymphoma, and multicentric Castleman disease (reviewed in Ganem, 2007; Mesri et al, 2010). KS is a vascular tumor of endothelial cells that affects elderly men in Mediterranean and African populations (endemic KS). However, with the onset of AIDS, a more aggressive and often lethal form of KS appeared. The virus infects the spindle cells of the skin, which are probably derived from lymphatic endothelial cells, and causes a proliferation of small red blotches over the entire body through a process of angiogenesis (Fig. 6–6). Body cavity-based or primary effusion lymphoma is a B-cell lymphoma, which occurs as malignant effusions in visceral cavities. These lymphomas are often coinfected with EBV, and in the setting of AIDS the tumors are extremely aggressive. Multicentric Castleman disease is a reactive lymphadenopathy characterized by expanded germinal centers and proliferation of endothelial vessels within involved lymph nodes. Although classified as a hyperplasia and considered nonneoplastic, it often precedes the development of non-Hodgkin lymphoma; it appears to be caused by viral secretion of IL-6. The KSHV tumors are usually found in immunosuppressed individuals and the virus is endemic in many populations without causing disease.
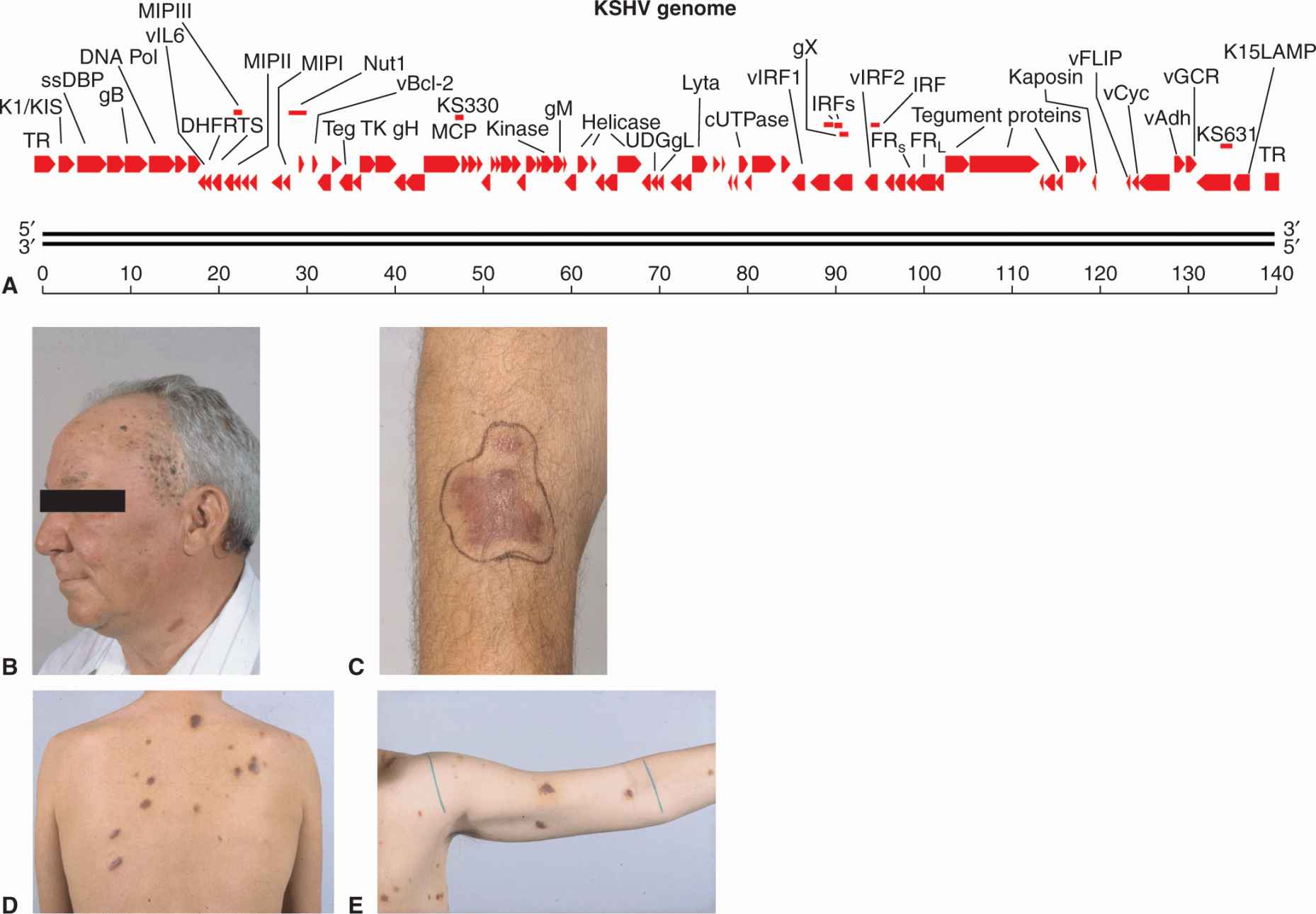
FIGURE 6–6 A) The linearized double-stranded DNA genome of KS virus showing important genes of the lytic cycle. The genome contains 87 open reading frames (ORFs) coding for latent proteins, reactivation proteins, and structural proteins. Host genes that help the virus evade immune surveillance and inhibit apoptosis have been acquired from chromosomes through a process of molecular piracy. These genes include vFLIP, vBcl-2, v-cyclin, interferon response factors (IRFs), and membrane cofactor protein (MCP), v-IL6, macrophage inflammatory protein (MIP) chemokines, LANA, and v-IL8R (vGCR). Genes specifying LANA, K1/KIST, and K15/LAMP maintain the viral episome in the latent state and are analogous to EBV proteins (EBNA1, LMP1, LMP2) with similar functions. B to E) Skin lesions associated with KS. Lesions present as red blotches on the skin caused by the infection of spindle cells. Angiogenesis occurs in the surrounding tissue. Panels B and C are examples of endemic or Mediterranean KS, while panels D and E illustrate epidemic or AIDS-related KS.
KSHV is a large, enveloped double-stranded DNA in the same family as the EBV. The double-stranded DNA genome is approximately 165 kbp in length and contains more than 85 open reading frames that encode virus-specific proteins. The virus can now be propagated in culture using immortalized dermal microvascular endothelial cells (Moses et al, 1999). The genome of KSHV encodes viral structural proteins, replicative enzymes, and host genes acquired through a process of “molecular piracy” that help the virus evade immune surveillance and inhibit apoptosis (see Fig. 6–6A). Many of the genes share homology with those of the EBV, and KSHV also specifies latency and lytic gene products.
The KSHV episome is maintained in the cell through use of latency-associated factors, including LANA (latency-associated nuclear antigen), which binds to viral DNA, inhibits p53-dependent transcription (Friborg et al, 1999), and binds to the retinoblastoma protein pRb (Radkov et al, 2000). It has similar properties to EBNA1 of EBV and large T antigen of SV40. LANA promotes latent replication of viral DNA, but suppresses transcription of lytic viral genes and deregulates some cellular genes. It inhibits p53 function and induces chromosomal instability (Si and Robertson, 2006) and also upregulates transcription of the proliferation gene survivin (Lu et al, 2009). LANA also induces IL-6 expression. Through its interaction with glycogen synthase kinase-3 beta (GSK-3β) it prevents the phosphorylation of β-catenin, allowing it to enter the nucleus and stimulate proliferative genes of the Wnt signal transduction pathway (see Chap. 8; Fujimuro et al, 2005). LANA can also block TGF-β to inhibit normal growth arrest and apoptosis (Di Bartolo et al, 2008). Another viral protein called vFLIP (viral FLICE inhibitory protein) can interact with and block TRAF-2 association with caspase 8 (FLICE) to inhibit apoptosis (Guasparri et al, 2006). vFLIP can also activate NF-κB signaling by interacting with the IKK complex (Bagneris et al, 2008). A viral homolog to cellular D-type cyclins (v-cyclin) can associate with cyclin-dependent kinase 6 (CDK6) and phosphory-late pRB, to activate E2F-induced transcription and promote G1-S cell-cycle transition and DNA replication (see Chap. 9, Sec. 9.2.3) reviewed in Verschuren et al, 2004). The v-cyclin/CDK6 complex is resistant to the CDK inhibitors p16, p21, and p27 and phosphorylates the p27 inhibitor to trigger its degradation. Transgenic mice expressing v-cyclin on a p53(–/–) background produced lymphoma with a 100% frequency, indicating that this viral protein probably has a role in tumorigenesis (Verschuren et al, 2002). The secreted viral IL-6 cytokine has 62% sequence similarity to its host cellular homolog. It binds to host cell receptors and stimulates cell growth, VEGF production, and transformation of spindle cells and B cells. By binding to the cellular receptor (gp130), it activates transcription activators (STAT1, -3, and -5) and can stimulate genes that block apoptosis, induce vascularization (VEGF), stimulate hematopoiesis, and prevent cell-cycle arrest caused by interferon-a (Chatterjee et al, 2002). Another viral homolog called viral G-protein–coupled receptor (v-GPCR) has sequence similarity to the cellular IL-8 receptor. Its signaling is constitutively active but also regulated by the binding of a variety of chemokines. It is known to activate MAPK, PI3K/Akt, NF-κB, and p38 pathways. It induces the expression of VEGF and cyclooxygenase-2 (COX-2) to stimulate angiogenesis and prostaglandin synthesis in endothelial cells. Transgenic mice expressing v-GPCR in hematopoietic cells developed angioproliferative lesions similar to KS in multiple organs (Yang et al, 2000; Montaner et al, 2003). Finally the kaposin family of viral proteins (A, B, C) are believed to mediate transformation (Muralidhar et al, 1998) and promote inflammation (McCormick and Ganem, 2005).
During latent infections, KSHV expresses two membrane proteins called KSHV immunoreceptor tyrosine activation motif (ITAM)-based signal transducer (K1/KIST) and latency-associated membrane protein (K15/LAMP). K1/KIST appears to have an analogous function to the LMP1 protein of EBV. It is structurally similar to the BCR and may act as a decoy molecule to prevent the reactivation of KSHV that occurs when the BCR is engaged. K1/KIST is a 46-kDa transmembrane glycoprotein that contains 2 SH2 motifs in its SH2 domain which can recruit nuclear factor of activated T cell (NFAT), syk, and vav kinases. It can also activate the PI3K/Akt/mammalian target of rapamycin (mTOR) pathway and promote cell survival (Tomlinson and Damania, 2004; Wang et al, 2006). K1/KIST can also upregulate NF-κB–dependent transcription (Samaniego et al, 2001). Like LMP1, the KIST protein has transformation properties in Rat-1 fibroblasts and primary human umbilical vein endothelial cells (Lee et al, 2005; Wang et al, 2006) Expression of K1/KIST results in secretion of VEGF (see Chap. 11, Sec. 11.4.1) and matrix metalloproteinase-9 (MMP-9) by endothelial cells. The other viral membrane protein, K15/LAMP, has similarities to both the LMP1 and LMP2A protein of EBV, and possesses up to 12 membrane-spanning domains; the cytoplasmic C terminus contains a src protein kinase-binding motif (SH2) with signal transduction properties. The constitutive phosphorylation of Y481 in the SH2 domain by Src, Lck, Yes, Hck, and Fyn protein kinases activates Ras/MAPK, NF-κB, JNK/SAPK pathways and results in the induction of IL-6, IL-8, and COX-2 (Brinkmann et al, 2003, 2007). The cytoplasmic tail of K15/LAMP also contains a TRAF binding domain that functions to upregulate NF-κB, AP-1, and ERK signaling. Overexpression of K15/LAMP seems to block the function of KIST, and prevents intracellular calcium mobilization. This protein also appears to antagonize BCR signaling and prevents reactivation of lytic infection from the latent state.
In addition to latency-associated proteins, KSHV has been shown to express up to 17 miRNA molecules from 12 genes located in the latency-associated region of the viral genome (Samols et al, 2007; reviewed in Ziegelbauer, 2011). Particular mRNAs that appear to be targeted by the viral miRNAs include osteopontin, thrombospondin-1, and plasticity-related gene 1 (Samols et al, 2007). Thrombospondin-1 has previously been reported to have tumor suppressor, antiangiogenic, and immune stimulator activities. These miRNAs probably function to maintain latency and help the virus avoid the cellular innate immune system.
As is the case with EBV, lytic replication can be triggered by chemical agents including butyrate and phorbol 12-myristate 13-acetate (PMA) that inhibit histone deacetylases and activate MAPK signaling, respectively. The viral protein Rta is a transactivator that regulates the switch from latency to a lytic infection that yields structural proteins and virus particles. It is a 691-amino acid protein that contains an N-terminal binding domain and a C-terminal transactivation domain. Rta requires interactions with cellular proteins like RBP-Jk (recombination signal binding protein Jk), AP-1, and OCT-1 in order to activate other viral transcriptional promoters. The activity of Rta can be suppressed and controlled by viral and cellular factors including interferon response factor 7 (IRF-7), KSHV Rta-binding protein (K-RBP), PARP-1, NF-κB, histone deacetylase 1 (HDAC1), KSHV bZIP, and LANA. However, Rta also has ubiquitin E3 ligase activity that can promote the degradation of these repressors (Yang et al, 2008; Yu et al, 2008). Viral infections respond to nucleotide analogs, such as ganciclovir and acyclovir, and these agents have been shown to reduce the symptoms of KS in HIV-1 infected patients. Highly active antiretroviral therapy (HAART) has reduced the incidence of AIDS-related KS. Radiation and chemotherapy with liposomal daunorubicin or doxorubicin and rapamycin have been found to be effective in treating KS. New drug treatments that target the VEGF and angio-genesis pathways are in clinical trials for KS (see Chap. 11, Sec. 11.7; Dittmer et al, 2012; reviewed in Mesri et al, 2010). Rapamycin is probably effective because it interferes with the dependence of KS cells on the PI3K/Akt/mTOR pathway. Small molecules that inhibit mTOR include sirolimus, tacrolimus, and everolimus and are in Phase II/III trials.
A variety of DNA viruses have been implicated in human cancers. HPVs, Merkel cell carcinoma virus, EBV, and KSHV all seem to produce well-defined malignancies. The role of SV40 as a human carcinogen is still pending, but it may play a role as a cofactor in the development of mesotheliomas and some bone cancers. Recently, torque teno (TT) viruses, members of a new class of single-stranded DNA viruses (Anelloviridae), have been suggested to play a role in some human leukemias and colon cancer based on PCR, epidemiological evidence, and an association with red meat (zur Hausen, 2012; zur Hausen and de Villiers, 2009). Further research may confirm the role of these and other viruses in human cancer.
6.3 RNA TUMOR VIRUSES
6.3.1 Retrovirus Life Cycle
Retroviruses are enveloped viruses of approximately 120 nm in diameter (reviewed in Goff, 2007). The outer envelope is a lipid bilayer that is derived from the plasma membrane of the host cell through a “budding” process. The virus contains viral glycoproteins that are encoded by the viral env gene. This glycoprotein is usually cleaved during the assembly process to yield 2 subunits, SU and TM, which remain tightly associated. The envelope surrounds a nucleocapsid core composed of capsid proteins derived from the viral gag gene. The core includes 2 identical single strands of viral RNA that are linked together in a dimer structure through their 5′ termini and encode the capsid proteins, protease, viral polymerase, integrase, and envelope proteins (gag-pro-pol-env; Figs. 6–7A and 6–8). In addition, some classes of retroviruses (eg, HIV and human T-cell leukemia virus [HTLV]) contain accessory genes that encode proteins that assist in mRNA synthesis and viral assembly, whereas others may contain an oncogene that contributes to cellular transformation. Bound to the RNA are several copies of the enzyme reverse transcriptase (RT) encoded by the viral pol gene.
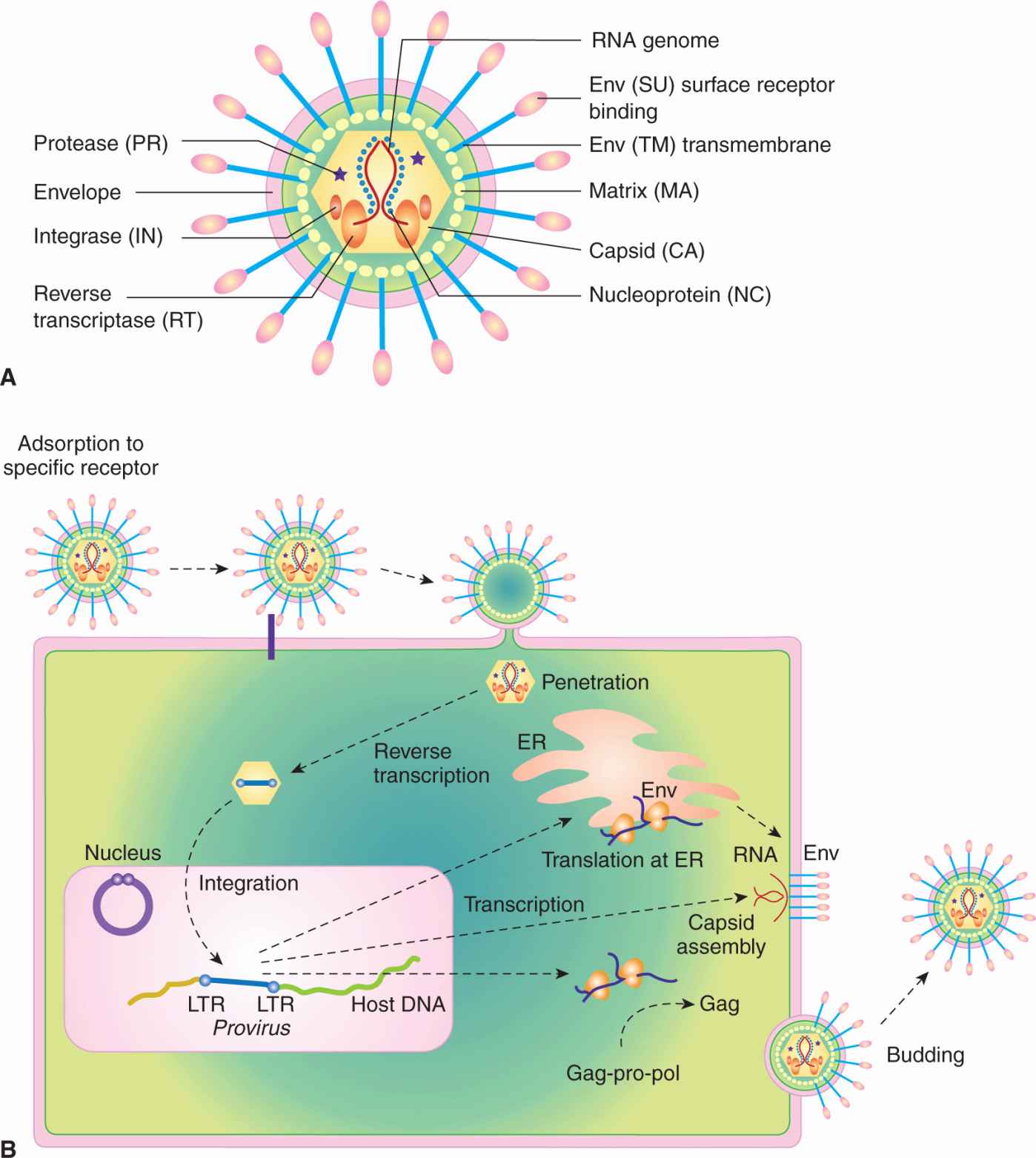
FIGURE 6–7 The structure and life cycle of a retrovirus. A) The virion possesses a membrane that contains an envelope protein that is cleaved into 2 subunits—surface receptor (SU) and transmembrane (TM) components. Beneath the envelope is a matrix protein (MA) that holds the membrane and the nucleocapsid together. The nucleocapsid consists of a capsid protein (CA), nucleoprotein (NC), reverse transcriptase (RT), integrase (IN), and protease (PR). The genome consists of 2 identical RNA strands. B) The retrovirus attaches to a specific receptor on the host cell. Its membrane subsequently fuses with that of the cell and the nucleocapsid is released into the cytoplasm. The single-stranded viral RNA genome is reverse-transcribed to a double-stranded DNA form, which has at its ends long terminal repeats (LTRs). The viral DNA migrates to the nucleus and integrates into the chromosomal DNA. The single viral transcript can form the genome for progeny viruses or can be processed and translated to generate viral structural proteins, gag or gag-pro-pol, and env. Gag and pol proteins are processed by the virus-associated protease to yield capsid (CA), and matrix (MA), polymerase (pol), and integrase (IN) proteins (see panel A). The envelope protein mRNA is translated at the rough endoplasmic reticulum and the protein precursor is cleaved to surface receptor (SU) and transmembrane (TM) subunits at the Golgi. Viral assembly occurs beneath the membrane of the host cell and the mature virus buds from the plasma membrane.
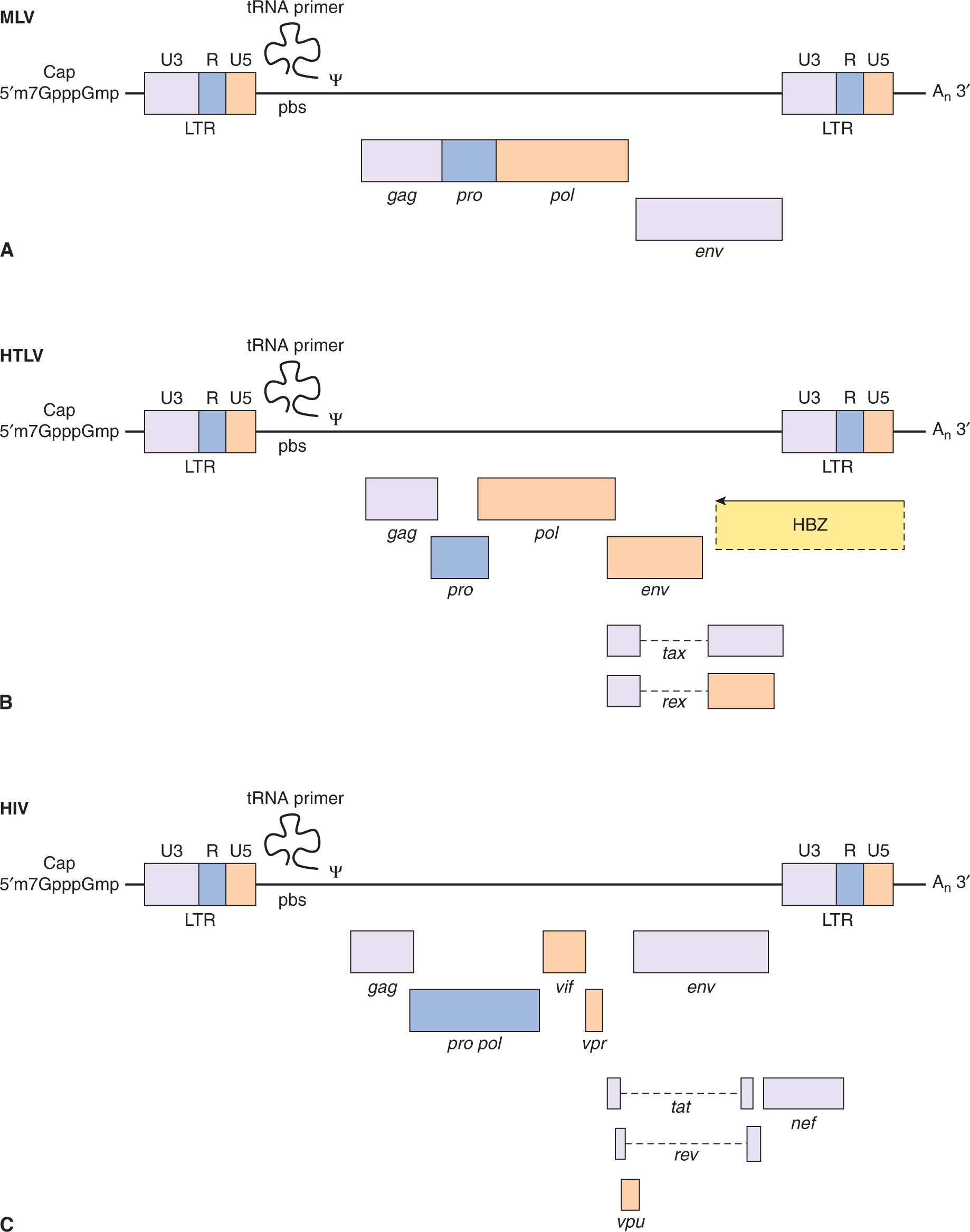
FIGURE 6–8 Genome organization of oncogenic retroviruses. A) Single-stranded RNA genome of Maloney leukemia virus (MLV) showing long terminal repeats (LTRs) and coding regions for gag, pol, and envelope (env) proteins. The RNA genome is 5′-capped and possesses a poly A sequence at its 3′ terminus. ψ represents the packaging signal with which capsid protein associates. B) In addition to the long-terminal repeats (LTRs) and the typical gag, pol, and env genes found in other replication-competent retroviruses, a novel region exists at the 3′ end of the HTLV genome encoding 2 regulatory proteins, Tax (transactivator) and Rex (regulator of expression). Three different mRNA species have been identified for HTLV-1. The full-length genomic mRNA encodes the gag and pol proteins and is also packaged into virions. A singly spliced mRNA encodes the env protein and the doubly spliced mRNA encodes Tax and Rex. HBZ is a protein translated from an antisense transcript derived from the 3′ end of the HTLV genome. C) The genome of HIV also codes for structural proteins, regulatory proteins (Tat and Rev), and accessory proteins (vpr, vpu, vif, and nef).
The life cycle of a retrovirus occurs through a sequence of discrete steps, which are illustrated in Figure 6–7B. Adsorption of the virus to a cell is mediated by an interaction between the envelope proteins of the virus and specific receptor molecules on the cell surface that leads to membrane fusion between the viral envelope and host cell plasma membrane. A fusion peptide found on the envelope protein mediates this event. Specificity at the level of virus adsorption accounts in large part for the restricted host and cell range of many types of viruses. HIV, for example, attaches to and enters a cell through the CD4 cell surface antigen; as a result, only CD4-positive cells are susceptible to infection by HIV. Host coreceptor molecules, such as the chemokine receptors CXCR4 and CCR5, can add further specificity at the level of membrane fusion and entry (Berger et al, 1999). Other receptors for retroviruses include the low-density lipoprotein receptor, tv-b locus (TNF family receptor), multiple membrane spanning amino acid transporter proteins, or the sodium-dependent phosphate symporter. Retroviruses infect mice, birds, reptiles, mink, cats, cows, and monkeys, but to a lesser extent humans, and can cause tumors. Compared to other viruses, these agents are fragile; overcrowding, close contact, and intimate behavior facilitates their transfer from one host to another. Retroviruses cause few cancers in humans and much of what we have learned about these viruses has been derived by isolates from mice and chickens. The protooncogenes targeted by these viruses have been instrumental in the elucidation and understanding of signal transduction pathways (see Chap. 8).
Once the virus is inside the cell, loss of the viral envelope produces a core particle that is permeable to entry by deoxyribonucleotides. The RNA is then converted to double-stranded DNA through the activity of the virus-encoded RT that occurs in a large cytoplasmic complex consisting of nucleocapsid, RT, integrase, and viral RNA. The +ve sense RNA genome serves as a template in this process and a large pool of deoxyribo-nucleotides may trigger this process. Reverse transcription occurs through the multistep process shown in Figure 6–7B. Initially, a small nucleotide repeat sequence at both ends of the viral RNA is extended to form a long terminal repeat (LTR) that is incorporated into the double-stranded DNA. These linear DNA molecules then cross the nuclear membrane. Simple retroviruses require the breakdown of the nuclear membrane during the normal process of cellular mitosis, whereas linear DNA of HIV-related viruses can cross the nuclear membrane of a nondividing cell. One or a few molecules of viral DNA integrate randomly into the host chromosomes in association with the viral integrase molecule. The integrated form of the virus is called the provirus.
Once integrated, the proviral DNA acts as a template for transcription. Although both LTRs are identical and contain promoter and enhancer sequences necessary for synthesis of viral RNA, the upstream LTR acts to promote transcription whereas the downstream LTR specifies termination. Between the LTRs are coding sequences for the gag, pro, pol, and env genes (see Fig. 6–8A). In a simple retrovirus, 2 transcripts are synthesized from the proviral DNA. A full-length genomic transcript serves as the mRNA for the synthesis of both gag and gag-pro-pol fusion proteins. This transcript can also be packaged into virus particles, and therefore acts as the genome of the virus. Gag, gag-pro-pol, and genomic RNA assemble beneath the env protein at the plasma membrane of the infected cell and the complete virion buds from the cell. Maturation of the viral particle occurs during the budding process as the viral protease (Pro) cleaves the gag precursor into matrix (MA), capsid (CA), nucleocapsid (NC), protease (Pro), RT (RT), and integrase (IN) polypeptides. This maturation is crucial to produce a fully infectious virion, and this protease is the target of inhibitors (indinavir or saquinavir) currently used to treat AIDS. Retroviral infections are surprisingly benign and do not have immediate cytopathic effects on the infected host cell. However, over the long-term, disease can become apparent as a result of the acquisition of host genes or insertional mutagenesis into the host chromosome by these viruses.
A widespread role for retroviruses in human cancers has never been documented. Only HTLV-1 and -2 have been implicated in the generation of acute adult T-cell leukemia/lymphoma (ATLL). The recent report that xenotropic mouse retrovirus (XMRV) played a role in human prostate cancer and chronic fatigue syndrome has been discounted as a laboratory contamination artifact caused by genetic recombination between 2 viral genomes carried in the germline DNA of mice during prostate cancer xenograft experiments (Sfanos et al, 2012). Despite the absence of disease associated with retroviruses in human, the human genome does contain genetic material derived from endogenous retroviruses. However, these endogenous retroviruses that are incorporated into human genomic DNA are defective and carry inactivating mutations and deletions in their coding regions and LTRs are silenced through hypermethylation. The human endogenous retroviruses do not cause disease and occasionally their promoters and enhancers are used to control host gene transcription (eg, pancreatic/salivary amylase; p63 tumor suppressor). Exogenous retro-viruses are inactivated by host restriction factors such as APOBEC3G (a cytidine deaminase), TRIM5α (binds a targets capsid for ubiquitination), Tetherin (prevents budding and release of the virion), and the innate immune system. Successful human retroviruses, such as HIV and HTLV, have developed accessory factors that appear to neutralize these restriction factors.
6.3.2 Acute Transforming Retroviruses
Transforming retroviruses can be separated into 2 major groups based on their different mechanisms of transformation. Some viruses contain a viral oncogene, and these have been termed acute or rapidly transforming viruses. More than 20 viral oncogenes have been identified; each of these has been found to have a counterpart in normal cells. Other viruses do not contain an oncogene and are referred to as chronic or slowly transforming viruses. Again, these cancer viruses affect mice and birds, and as yet have not been implicated in human tumorigenesis.
Acute transforming viruses are almost always replication-defective as a result of replacement of viral sequences required for replication with host-derived oncogene sequences. For example, MC29 is a defective virus containing v-myc and is missing all of the pol gene and parts of gag and env. As a result, these transforming viruses require the presence of replication-competent helper viruses that assist in viral replication and assembly by supplying the necessary viral gene products. Because viral oncogenes come under the control of the efficient retroviral promoter present on the LTRs and are no longer tightly regulated by cellular mechanisms that normally act on the natural promoter, these genes can be expressed at inappropriately high levels. The viral oncogenes are frequently mutated because of the poor fidelity of retroviral replication, and often contain point mutations, deletions, substitutions, and insertions when compared with the protooncogenes from which they are derived. In addition, viral oncogenes differ from protooncogenes in that they do not contain intron (ie, noncoding) sequences. Retroviruses containing oncogenes can transform cells in culture after several days and can induce leukemias and sarcomas in infected animals relatively quickly. Expression of the v-onc gene transforms every infected cell. Consequently, polyclonal tumors develop from many different infected progenitor cells. Table 6–1 summarizes examples of oncogenes and their cellular counterparts that have been transduced by acute transforming viruses. Viral oncogenes include growth factors (v-Sis), growth factor receptors (v-erbB), intra-cellular tyrosine kinases (v-src, v-fps, v-fes, v-abl), serine/threonine kinases (v-Akt, v-Raf), G proteins (H-ras, Ki-ras), transcription factors (v-myc, v-erbA, v-fos, v-jun), and many other protein families (see Chap. 7).
TABLE 6–1 Oncogenes recovered in transducing retroviruses.
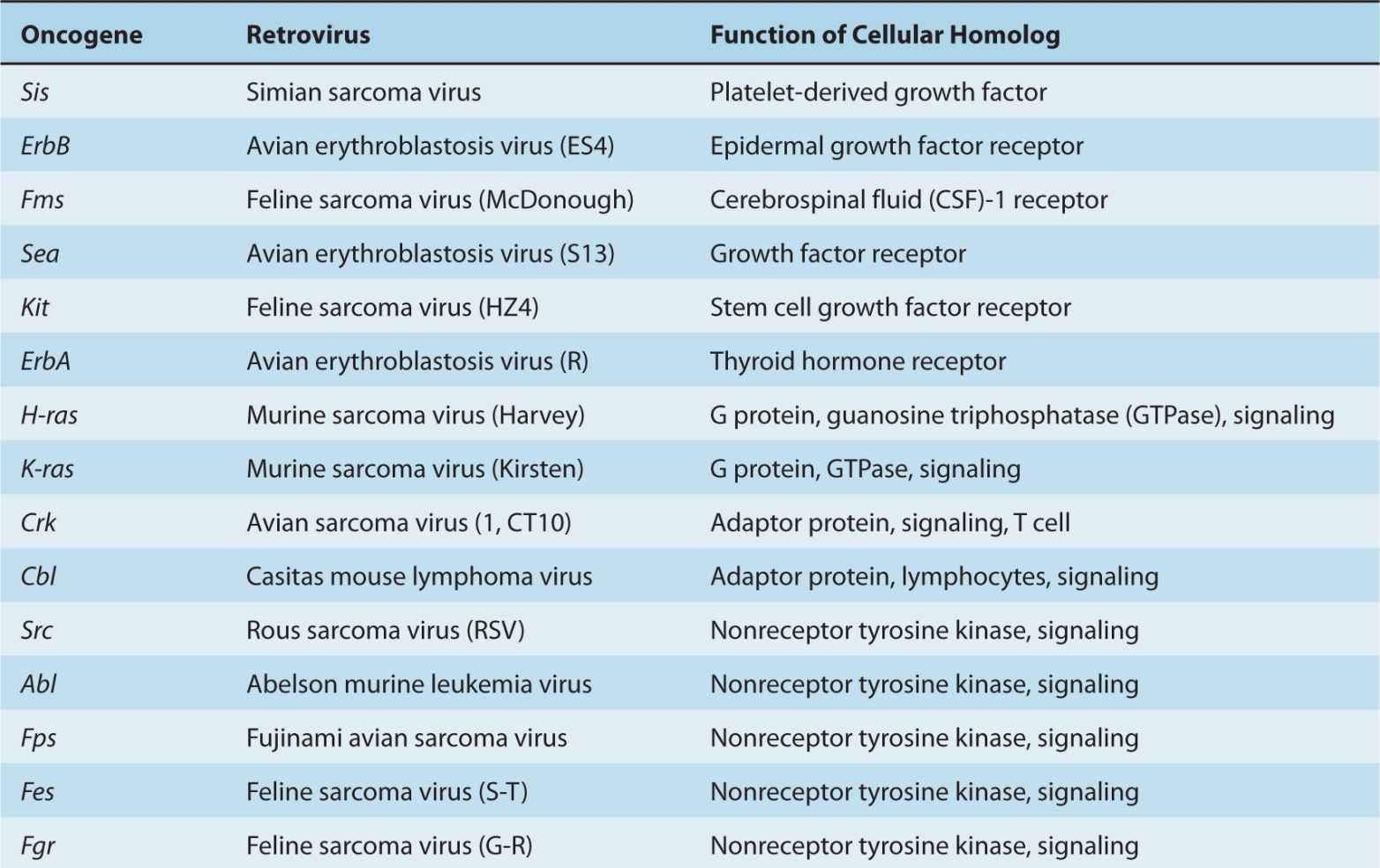

6.3.3 Chronic Tumor Retroviruses
The replication-competent chronic tumor viruses do not contain viral oncogenes but transform infected cells through a mechanism known as insertional mutagenesis, in which proviral integration leads to the aberrant activation or sometimes inactivation of adjacent cellular genes. Protooncogenes may be activated by LTR promoter insertion, LTR enhancer insertion, viral poly-A site insertion that stabilizes mRNA, or viral leader insertion that increases and stabilizes mRNA. In other instances a cellular gene may be activated or inactivated by insertion of the retrovirus in the middle of a cellular gene depending upon the arrangement of exons. Several of the protooncogenes identified initially as progenitors to transducing retroviral oncogenes (c-erbB, c-mos, c-myb, c-myc, c-H-ras, c-K-ras, c-fms, c-fli1) have also been identified through insertional mutagenesis. Cytokines regulating cell growth (IL-2, IL-3, IL-10) are also activated by retroviral integration in several animal species. Usually more than 1 insertional mutation event is required to produce tumors in animals.
Avian leukemia virus (ALV) is a typical slow-acting retro-virus. In ALV-induced B-cell lymphomas, malignant clones transformed by these viruses contain proviruses integrated in the vicinity of the c-myc gene. In many tumors, the provirus is integrated upstream of c-myc and in the same transcriptional orientation. In such cases the 3′ LTR, which normally acts to terminate viral transcription, promotes transcription of c-myc sequences. The resulting hybrid RNA transcripts contain both viral and c-myc sequences and are present at levels 30- to 100-fold higher than that of c-myc RNA in normal tissues. Such c-myc transcripts appear to encode a normal c-myc protein. This mechanism is called promoter insertion. In other tumors, the provirus is integrated upstream of c-myc but oriented in the transcriptional direction opposite to that of the gene, or it is integrated downstream of the gene. The strong enhancer properties of the LTRs are then believed to be responsible for activation of c-myc transcription, and this mechanism of transformation is known as enhancer insertion. The majority of B-cell lymphomas induced by ALV contain proviral sequences adjacent to c-myc, but the myc oncogene requires the cooperative function of a second oncogene. Because integration adjacent to c-myc is a random, rare event and secondary genetic events are required for tumor progression, ALV-induced leukemia may arise slowly and is clonal in origin.
Other examples of transformation by chronic tumor viruses include insertion of the mouse mammary tumor virus (MMTV) next to the wnt-1/int-1 or hst/int-2 genes leading to mammary tumors in mice. The hst/int-2 protein belongs to the FGF family and can be upregulated in KS, stomach cancer, and teratocarcinomas, as well as in mouse mammary tumors. The Lck gene, normally found in T cells, NK cells, and some B cells, was found to be upregulated in lymphoma cell lines derived from thymomas of murine leukemia virus-infected mice. Other oncogenes that have been found as a result of proviral insertions include Ahi-1, Evi-1, Evi-2, Fli-1, Mlvi-1, Mlvi-3, and Pvt-1.
Proviral insertion may disrupt or alter the protein-coding sequence of resident cellular genes. For example, in ALV-induced erythroblastosis, proviral insertions commonly map to a small region in the middle of the EGFR gene (c-erbB; see Chap. 7). The resulting transcripts contain viral gag and env sequences fused to c-erbB sequences. The amino acid sequence predicted from these hybrid transcripts contain amino acids encoded by gag and env fused to carboxy-terminal amino acid sequences encoded by c-erbB. Thus, expression of an altered, truncated EGFR molecule appears necessary for the development of ALV-induced erythroblastosis.
6.3.4 Human T-Cell Leukemia Virus
HTLV-1 and HTLV-2 are the only retroviruses that are known to lead directly to cancers in humans (reviewed in Lairmore and Franchini, 2007; Matsuoka and Jeang, 2011). HTLV-1 by itself can cause ATLL, a rare but virulent cancer that is endemic in southern Japan, the Caribbean, northern South America, parts of Africa, and the southeastern United States. The virus has infected approximately 20 million people worldwide and over a prolonged period of 20 to 40 years it can produce ATLL in 1% to 5% of infected individuals. Transmission of this disease may occur through blood transfusion, breast feeding, and sexual intercourse. Nucleotide sequence determination of the viral genomes has shown that the Japanese and American isolates were closely related strains of a single retrovirus now called HTLV-1.
There are 4 molecular subtypes of HTLV-1 (A to D), which reflect the geographical locations from which the virus is isolated. The virus exhibits little genetic variation and there is an approximately 98% homology between different HTLV-1 strains. HTLV-1 has since been recognized as an agent that causes ATLL. In some chronically infected patients the virus can also cause a neurodegenerative disease called HTLV-associated myelopathy (HAM) or tropical spastic paraparesis (TSP). HTLV-2 is much less common than HTLV-1 and a convincing role of this virus in human disease has not been demonstrated, although a few patients have exhibited neurological disease.
Unlike the common oncogenic retroviruses of animals, HTLV-1 does not carry a host-derived oncogene and does not activate cellular protooncogenes by insertional mutation. HTLV-1 is believed to initiate a multistep process leading to ATLL. These steps include an asymptomatic carrier state, preleukemic state, chronic/smoldering ATLL, lymphoma type, and acute ATLL. Both HTLV-1 and HTLV-2 can immortalize human peripheral blood T cells in vitro, as well as T cells from monkeys, rabbits, cats, and rats. The cellular receptor for the virus is the glucose transporter protein GLUT-1. Entry into the host cell and cell to cell fusion require the participation of coreceptors, which include cell adhesion molecules (ICAM-1, ICAM-3, V-CAM, integrins). The virus can infect either CD4+ or CD8+ T cells, but transformed cells from ATLL patients are usually CD4+ and only occasionally CD8+. Following infection of T lymphocytes by HTLV-1, surface IL-2 receptors are upregulated, and the provirus is found randomly integrated into the cellular genome. The infected cell population undergoes a transient polyclonal expansion followed by a latency period that is variable in duration; it can be as short as a few years if infection occurs in adulthood or as long as 40 years if infection occurs in infancy. Not all individuals infected by the virus develop ATLL, and the latent state may be maintained by immuno-logical clearance of the infected cells. An infected individual will have approximately a 1% chance of developing ATLL over a lifetime. If a patient progresses to pre-ATLL, 50% of patients still have the chance of undergoing spontaneous regression. When ATLL is clinically evident, however, all the leukemic cells in a patient have a common proviral integration site in the host DNA, but no 2 patients have the same integration site. These observations suggest that HTLV-1 is derived from a single, infected, progenitor cell but that subsequent genetic events are required to induce ATLL.
Figure 6–8B illustrates the genome of HTLV-1. In addition to gag, pol, and env genes, HTLV encodes accessory proteins, Tax, Rex, p12, p13, p30, and the recently discovered HBZ, which is expressed from the antisense strand. The Tax protein is the transforming component of HTLV-1; it is critical for viral replication and functions as a transcriptional coactivator of viral and cellular gene expression (Matsuoka and Jeang, 2011). Tax protein contacts a GC-rich region in the U3 promoter region of the proviral DNA and recruits cellular transcription factors that include CREB, CREB binding protein (p300/CBP), which is a histone acetylase, the CBP-associated factor (PCAF), and transcription factors IIA, IIB, IID (Fig. 6–9A). The Tax protein is a multifunctional protein composed of several domains (Fig. 6–9B). In addition to regulating transcription, Tax upregulates the activity of NF-κB to increase the expression of IL-2, IL-15, IL-2R, and IL-15R. Tax does this by binding to and activating the IKK complex, which targets the NF-κB inhibitor (IkB) for ubiquitination and degradation, allowing NF-κB to enter the nucleus and trigger the expression of many cytokines (Fig. 6–9C). Mutations in Tax have shown that both CREB and NF-κB activities are required for efficient transformation. Cellular genes that are responsive to transcriptional activation by Tax include IL-2, the α subunit of the IL-2 receptor, granulocyte-macrophage colony-stimulating factor, protooncogenes c-sis and c-fos, and proliferating cell nuclear antigen (PCNA). Tumor cells from ATLL patients as well as T lymphocytes transformed in culture with HTLV-1 display an activated T-cell phenotype characterized by expression of IL-2 cell surface receptors and cell adhesion molecules such as ICAM-1. Nonhuman and rodent models for HTLV-1 exhibit little disease progression. However, transgenic Tax expression under control of the granzyme B promoter produced large granular lymphocytic leukemia in mouse T cells (Grossman et al, 1995). ATLL-like T-cell leukemia/lymphoma was produced in mice with Tax driven by the Lck promoter (Hasegawa et al, 2006; Ohsugi et al, 2007). At the molecular level, overexpression of Tax can lead to inhibition of DNA repair as a result of overexpression of the PCNA processivity factor (see Chap. 5, Sec. 5.3). Tax can also bind to PI3K and activate Akt (PKB), which both promotes cell survival and inhibits apoptosis (Sun and Yamaoka, 2005). In addition Tax can accelerate cell cycle progression by binding and stabilizing cyclin D3, cyclin D2, and Cdk4 (Marriott and Semmes, 2005; see Chap. 9, Sec. 9.2.2). It can also inactivate retinoblastoma protein (Rb) and p53 tumor suppressor (Miyazato et al, 2005). The Tax protein is also believed to be responsible for the “flower-like” polynuclear lymphocytes that characterize ATLL by interfering with mitotic spindle checkpoint control (Liu et al, 2005; Chi and Jeang, 2007). Thus, expression of the tax gene has the potential to perturb many normal cellular functions leading to malignancy.
FIGURE 6–9 Molecular domains and functions of HTLV-1 Tax protein. A) Tax protein contacts a GC-rich region in the U3 promoter region of the proviral DNA LTR and recruits cellular transcription factors that include CREB, CREB binding protein (CBP/p300), which is a histone acetylase, the CBP-associated factor (PCAF), and transcription factors IIA, IIB, IID. B) Functional domains of Tax showing CREB binding site, nuclear localization sequence (NLS), p300/CBP binding region, NF-κB–activating domain, and PCAF binding region. C) Tax upregulates expression of IL-2R, AP1, p21WAF/CIP1, and Bcl-xL, and activates NF-κB, which augments the expression of hundreds of genes, including IL-2, IL-15, IL-2R, and IL-15R. It blocks the functions of p53 and p16INK4A.
Rex is essential for viral replication and acts post-transcriptionally to upregulate the levels of virion proteins to assure the production of infectious virus. The protein Rex binds to recognition sequence near the 3′ ends of genome-length unspliced RNA and partially spliced mRNAs, allowing them to be exported from the nucleus to the cytoplasm, instead of being subjected to splicing or degradation. The Rex complex binds to the nuclear export factor exportin-1 (CRM-1) and releases the unspliced RNA in the cytoplasm for translation or packaging. This allows for production of viral structural proteins (gag, pol, pro, env) and packaging of the RNA genome. Rex is the regulatory switch between latent and productive phases of the HTLV-1 life cycle and favors the production of virus particles (Baydoun et al, 2008; reviewed in Younis and Green, 2005). Rex also stabilizes the mRNAs for IL-2, IL-2R, the kinase FynB, vascular cell adhesion molecule (VCAM)-1, and leukocyte function-associated antigen (LFA)-3. The p12 protein resides in the endoplasmic reticulum and Golgi and interacts with the chaperones calnexin and calreticulin to increase intracellular Ca++ levels, which results in the dephosphorylation and activation of the transcription factor NFAT that regulates proliferation and differentiation of T lymphocytes. P12 can also bind the subunits of IL-2R, the transcription factor STAT5, and the MHC-I heavy chain. P30 binds the mRNAs for Tax and Rex and tethers them inside the nucleus to inhibit their protein synthesis, thus maintaining the virus in a latent state. It also promotes cell survival and inhibits apoptosis. HBZ is a protein that is translated from an antisense transcript derived from the 3′ end of the HTLV-1 genome. The protein contains a leucine zipper domain that can interact with CREB, CREB-2, CREM-Ia, ATF-I, c-Jun, JunB, JunD, and NF-κB transcription factors. It acts to suppress Tax-mediated transactivation of viral transcription at the 5′-LTR and the Hbz RNA promotes ATLL cellular proliferation. Tax is needed to initiate transformation, whereas HBZ is required for cell proliferation and maintenance of the transformed phenotype late in ATLL when Tax expression diminishes (Satou et al, 2006; Lemasson et al, 2007).
The mean survival time for aggressive ATLL without treatment is less than 1 year. Standard treatment usually starts with cytostatic chemotherapeutic agents such as vincristine, cyclophosphamide, doxorubicin, ranimustine, vindesine, etoposide, carboplatin, and prednisone (reviewed in Goncalves et al, 2010; Matsuoka and Jeang, 2011). Alternatively, nucleo-side analogs and topoisomerase inhibitors have been used, resulting in limited improvement. Chemotherapy can yield a 3-year survival rate of about 25%. The major obstacle in therapy is multidrug resistance of the ATLL cells coupled with the patients’ immunodeficient state. Complications from opportunistic fungal, viral, protozoal, and bacterial infections can worsen the prognosis. Allogeneic stem cell transplantation is effective in ATLL patients, with a 3-year survival rate of about 35%. Cell-mediated immunity against HTLV-1 is augmented in 30% to 40% of the surviving patients, leading to complete remission. A humanized antibody against CCR4 that is found on T-regulatory (T-reg) cells has also been effective in treating ATLL patients. NF-κB and histone deacetylase inhibitors may offer a new direction in therapy (Rauch and Ratner, 2011). The virus-associated neurological disease, HAM/TSP, is generally untreatable and it responds poorly to treatment with corticosteroids, plasmapheresis, interferon, and antiretroviral drugs.
6.3.5 Human Immunodeficiency Virus
HIV is a member of the retrovirus family and has been classified as a lentivirus. The RNA genome of HIV encodes core (gag), polymerase (pol), and envelope (env) gene products in addition to regulatory and accessory proteins (Tat, Rev, Nef, Vif, Vpr, and Vpu) that play key roles in the pathogenesis of the viral infection (see Fig. 6–8C). Tat is a transactivating protein of the LTR promoter and Rev interacts with and directs the export of unspliced viral mRNAs to the cytoplasm and favors synthesis of viral structural proteins. Vif increases viral infectivity of certain cell types and Vpr promotes nuclear localization of proteins from the cytoplasm. Vpu enhances the release of virus from the cell. Nef is critical in maintaining high virus loads, increases circulating virions, and down-regulates surface expression of CD4 and MHC I (see Chap. 21, Fig. 21–1). The virus initiates infection by binding to the CD4 receptor on T-cells and macrophages, and penetrates into the host cell through use of chemokine coreceptors and a process of membrane fusion. Viral replication occurs as described in Figure 6–7B. HIV causes immune suppression through destruction of lymphoid tissue and depletion of CD4+ T cells. Combinations of antiretrovirus nucleotide analogs and viral protease inhibitors are used as a short-term therapy to suppress plasma viral loads. This treatment is known as HAART.
Although HIV does not cause cancer directly, it is an immunosuppressive virus that can lead to the appearance of several types of HIV associated malignancies during late-stage infections (Casper, 2011). These AIDS-related malignancies are the result of reactivation of infections with HPVs, EBV, and human herpesvirus 9 (KSHV; see Sec. 6.2.6). The cancers include smooth muscle sarcomas (leiomyosarcoma), Hodgkin disease, NPC, AIDS-related lymphomas (non-Hodgkin lymphoma, Burkitt lymphoma, primary central nervous system lymphoma), cervical cancer, anal carcinomas, HCC, lung cancer, squamous cell neoplasia, primary effusion lymphoma, and KS. HAART can be effective in treating less-aggressive cancers, but tumor-specific therapy may be indicated. With the significant improvement in morbidity, mortality, and life expectancy of AIDS patients, non-AIDS–defining malignancies have been on the rise (Deeken et al, 2012). These include cancers of the lung, breast, colon, anus, kidney, and skin, which develop over longer periods of time following successful HAART treatment. A major challenge to oncologists is how to administer chemotherapy safely and effectively to patients on retroviral therapy.
6.3.6 Hepatitis C Virus
HCV is a member of the family of viruses that includes yellow fever virus and dengue fever virus. Following the discovery of HBV in 1965, and the characterization of hepatitis A virus in 1973, it became apparent that other agents responsible for blood-borne hepatitis also existed and caused non-A, non-B hepatitis. This led to the discovery of the HCV. The HCV virion is approximately 50 nm in diameter and consists of an envelope that contains the 2 viral glycoproteins, E1 and E2. Surrounding the positive-sense, single-stranded RNA genome is the core or nucleocapsid protein. The genome encodes the structural proteins (core, E1, E2) and nonstructural proteins (p7, NS2, NS3, NS4A, NS4B, NS5A, and NS5B) which are required for viral replication (Fig. 6–10). There are 7 known genotypes of this virus that reflect their geographical distribution. HCV replication is slow and inefficient and the virus establishes persistent viral infections that are not cytotoxic. Two major barriers have hampered research in the HCV field: the virus cannot be easily propagated in cultured cells, and the previous lack of a small animal model in which to propagate and study the immunology of this pathogen and evaluate antiviral agents. Chimpanzees are the only primates besides humans that support the replication of HCV. An important breakthrough was the development of a severe combined immunodeficiency (SCID) urokinase transgenic mouse model into which human liver cells have been introduced. This murine model promises a useful tool with which to study antiviral agents and production of the virus in vivo (Mercer et al, 2001). Another major discovery was reported when a single strain of Japanese fulminant hepatitis C (JFH-1) was found to grow and produce virus particles in a hepatoma cell line (Huh 7.5) (Wakita et al, 2005). This cell line has allowed researchers to study the interaction of HCV with the innate immune system, elucidate viral receptors on the cell, and study viral RNA synthesis in association with intracellular membrane structures.
FIGURE 6–10 RNA genome and polyprotein of HCV showing structural and nonstructural viral proteins. The single open reading frame of the genome translates into a polyprotein that is processed by host and viral proteases into 10 proteins. These include structural components for capsid (C) and 2 envelope/membrane proteins (E1, E2). A viral pore protein (p7) appears to be important during virus assembly. Nonstructural proteins include a metalloprotease (NS2), protease-helicase (NS3/NS4A), RNA polymerase (NS5B), and replication factors whose functions are being defined (NS4B, NS5A). Cleavage sites for host proteases are shown in blue, the NS2 metalloprotease in red, and the NS3/NS4A serine protein in black.
HCV is blood borne and is spread through transfusions, intravenous drug use, tattoo parlors, multiple use of needles for vaccination, and sexual contact. The acute phase of hepatitis C is often mild and undiagnosed in both adults and children. Malaise, nausea, right upper quadrant pain, and dark urine may appear in about one-third of patients. Liver transaminase (alanine aminotransferase [ALT]) levels in the blood may increase and jaundice is only occasionally evident. However, HCV goes on to establish persistent viral infections and chronic disease in 60% to 80% of these patients leading to cirrhosis and HCC. Damage to the hepatocytes in the liver occurs through the effects of cytotoxic T cells and the action of Fas ligand and perforin on these target cells. The virus undergoes genetic variation in the face of a mounting humoral response and a region of the E2 proteins called the hypervariable 2 (HVP2) region, is particularly prone to muta-genesis. Low-grade persistent viral infections appear to survive in the face of a vigorous immune response against the virus: the virus may survive immune surveillance through the generation of mutants that escape T-cell cytotoxicity (Cooper et al, 1999; Wong et al, 2001). HCV can also induce anergy in both CD4+ T-helper cells and CD8+ CTL cells (Lechner et al, 2000), which prevents clearance of virus and contributes to persistent viremia. Alcohol abuse in hepatitis C patients can accelerate the progression and severity of the disease. This could be related to alcohol induced immunosuppression or the production of proinflammatory cytokines that lead to cytotoxicity and apoptosis.
Approximately 80% of patients with HCC also have cirrhosis. The genome of HCV is not transcribed to DNA and does not integrate into the host chromosome. The HCV core protein itself has oncogenic potential based on studies with core transgenic mice. These mice can exhibit liver steatosis (lipid droplets) and develop HCC after approximately 16 months (Lerat et al, 2002). Chronic liver inflammation caused by the HCV infection is believed to be an important carcinogenic factor. Hepatocellular death as a result of chronic inflammation and subsequent regeneration of hepatocytes can yield mutagenesis in the liver. At the molecular level, core protein has been reported to activate the JNK signal transduction pathway (Tsutsumi et al, 2003), inhibit the suppressor SOCS-1 (Miyoshi et al, 2005), upregulate NF-κB activity (Tai et al, 2000), and inactivate p53 (Yin et al, 1999). Other proteins also influence host cells but whether the gene products of HCV contribute to liver cancer remains unknown.
HCV can also cause B-lymphocyte proliferative disorders, including mixed cryoglobulinemia and non-Hodgkin lymphoma (NHL) (Ferri et al, 1994). Approximately 20% to 50% NHL patients in Italy exhibit HCV infections (Ferri et al, 1997). Other studies in Japan and the United States also indicate a high prevalence of HCV in cases of NHL (Zuckerman et al, 1997). Infection of B cells with HCV can yield a mutator phenotype which is characterized by increased mutations in immunoglobulin genes, protooncogenes, and tumor-suppressor genes (Machida et al, 2004). These genes include β-catenin, BCL-6, and p53. The mutations can be induced through double-stranded DNA breaks caused by activation of error-prone DNA polymerase ζ, polymerase t, and activation-induced cytidine deaminase. This process could be induced by interaction of E2 protein of HCV with the CD81 virus core-ceptor, aggregation of CD19 and CD21 and stimulation of the BCR signal transduction pathways (Weng and Levy, 2003). HCV core, NS3, and NS5A proteins have also been reported to induce reactive oxygen species, increase NO production, and cause chromosome translocations (Lai, 2002; Machida et al, 2006).
Previous treatment for hepatitis C involved the administration of pegylated interferon in combination with ribavirin over a period of 1 year and produced a sustained antiviral response in about half of infected patients, depending largely on the genotype of HCV being treated. The recent launch of NS3/4A protease inhibitors (telaprevir, boceprevir) and imminent release of NS5B polymerase inhibitors promises to cure HCV-related hepatitis for those patients that can afford treatment (Sarrazin et al, 2012). Prophylactic vaccines for hepatitis C are under development, but progress is slow and limited by the hypervariable nature of the virus (Houghton, 2011).
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


