Oncogenes and Tumor-Suppressor Genes
7.1 INTRODUCTION
Cancer is fundamentally a genetic disease. It results in expansion of a cellular population that invades and destroys surrounding organs and tissues and gains the ability to spread throughout the body. The second half of the 20th century inaugurated a steady stream of breakthroughs in the field of cancer research, largely spurred by an explosion of technologies enabling the analysis of tumors at a molecular level. Recent dramatic advancements in low-cost high-throughput sequencing of cancer genomes and the development of highresolution genome-wide profiling of the changes in gene copy number and structure are enabling detailed mapping of the genetic events associated with cellular transformation from nonmalignant to malignant cell.
The emerging comprehensive surveys of cancer genomes are uncovering the complexity of the disease and are also beginning to guide the development of highly targeted anticancer treatments based on discrete molecular features. Importantly, these efforts are revealing the extreme genetic heterogeneity that exists amongst tumors with similar histopathology (see also Chap. 13, Sec. 13.2.2). Although a given tumor may harbor hundreds of mutations in protein coding regions, perhaps only 10 to 15 can truly be considered to be “driver” mutations that confer a selective growth advantage to transformed cells. The remaining “passenger” mutations result from the genetic instability of cancer cells without actively contributing to oncogenesis. Distinguishing between the driver and passenger mutations is critical for the identification of therapeutic targets and the development of tailored treatment regimens with the promise of maximizing anti-cancer activity whilst minimizing side effects (Fig. 7–1) (Carter et al, 2009; Carter et al, 2010).
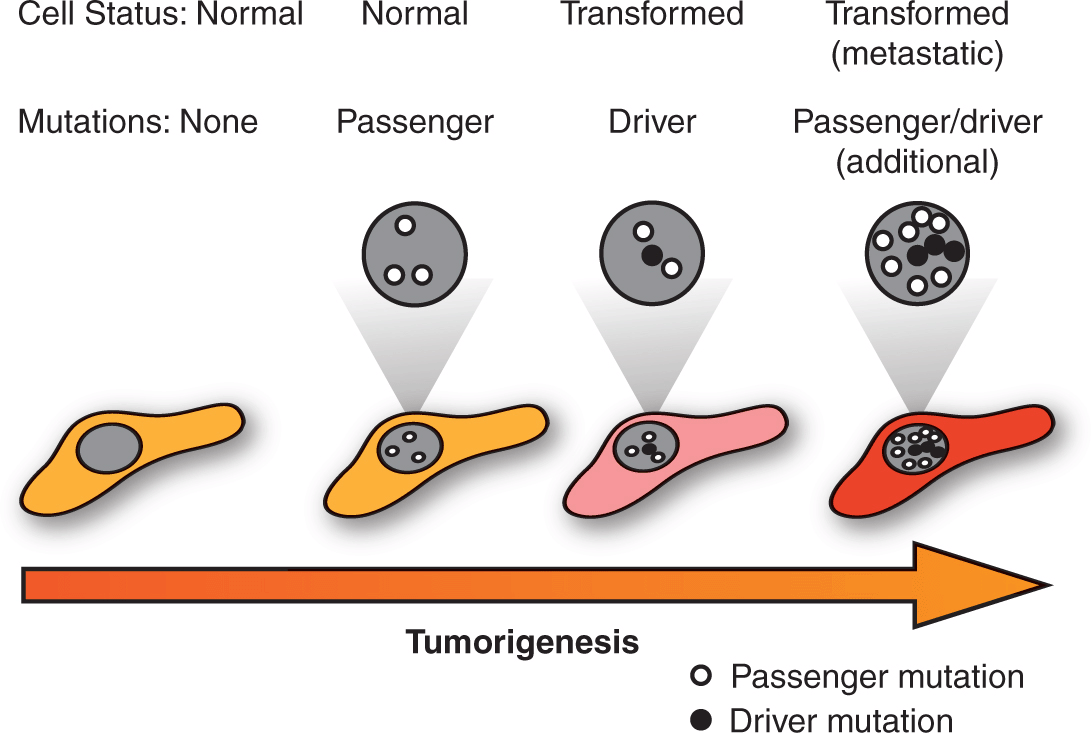
FIGURE 7–1 Passenger and driver mutations. Tumor sequencing has revealed that transformed cells acquire a genetic landscape marked by the accumulation of numerous mutations. Amongst these genetic changes, however, are a much more limited subset of driver mutations that actively impart oncogenic properties, with the remaining passenger mutations representing inert changes that are neither selected for or against during tumorigenesis. Distinguishing between driver and passenger mutations is a critical research objective as the former represent potential therapeutic targets.
7.2 THE GENETIC BASIS OF CANCER
7.2.1 Historical Perspective
The earliest recorded cases of cancer were documented in ancient Egypt around 1600 BC and described 8 cases of breast cancer along with a cauterization technique used to treat the disease. The term cancer itself was coined by the father of medicine, Hippocrates, who also advanced the theory that the origins of the disease resided in an excess of “black bile,” 1 of the 4 constituent fluids the ancient Greeks believed made up the human body. Remarkably, this humoral theory persisted as the dominant explanation for cancer for nearly 2 millennia thereafter. The genesis of understanding cancer as a genetic disease is widely traced to experiments carried out more than a century ago, the implications of which would go largely unappreciated for decades afterward. In 1902, the German biologist Theodor Boveri manipulated the structure of sea urchin chromosomes and was able to correlate malignant cellular growth with genomic abnormalities (Balmain, 2001). Strikingly, on the basis of these findings, he presciently speculated on the existence of cell-cycle checkpoints, oncogenes, and tumor suppressors, which are now known actors in oncogenesis. However, the first correlation between chromosomal defects and cancer in humans was not reported until 1960, with the discovery of the chronic myeloid leukemia (CML)-associated Philadelphia chromosome, generated by a gene translocation (Goldman and Melo, 2008).
7.2.2 Transforming Retroviruses
Another landmark breakthrough in the field of cancer research came in 1911, when Peyton Rous demonstrated that cell-free filtrates isolated from chicken sarcomas, later determined to contain retroviruses (see Chap. 6, Sec. 6.4), were capable of inducing tumor growth when introduced into healthy birds (Rous, 1911). Rous carefully documented the various characteristics which these tumors shared with the sarcomas, such as morphological appearance and the capacity to metastasize and invade distant tissues. Although some time would pass before his work gained widespread appreciation, his experiments ultimately led to the isolation of viral oncogenes from a number of transforming tumor viruses. In 1966, Rous was awarded the Nobel Prize in Medicine or Physiology, 55 years after the publication of his seminal work.
7.2.3 The Knudson Two-Hit Hypothesis
During the 1950s and 1960s, some researchers sought to explore the genetics of cancer by employing mathematical modeling to approximate the number of mutations required to induce neoplastic transformation, based upon the frequency of cancer incidence and estimated mutation rates. In particular, these models sought to explain the synergistic increase in tumor formation observed in mice subjected to repeated exposure to carcinogens, as well as the age-dependent acceleration in the onset of human cancers. In 1953, Carl Nordling estimated that roughly 7 mutations were involved in cancer progression, based upon his analysis of the age of cancer onset in several Western countries (Nordling, 1953). Peter Armitage and Richard Doll reported a similar conclusion the following year, before revising their estimates down to 2 oncogenic mutations for tumor formation in 1957 (Armitage and Doll, 1954; Armitage and Doll, 1957). Alfred Knudson elegantly reiterated these models of multistage cancer progression in 1971, in the form of his now famous “two-hit” hypothesis (Knudson, 1971). Knudson believed that the mutational requirements of cancer progression could be delineated by comparing the incidence rates of inherited and sporadic forms of retinoblastoma. He concluded that individuals who had inherited 1 mutant allele presented the disease at a frequency consistent with a single somatic mutation, whereas individuals who had not inherited a mutant allele exhibited an age-onset pattern consistent with 2 mutations. The onset of hereditary retinoblastoma represents the archetypal example of loss of heterozygosity (LOH) at a tumor-suppressor locus, generated by the inheritance of a single mutant allele, encoding an inactive protein, along with a copy of the wild-type allele. In the heterozygous state, sufficient functional tumor suppressor is expressed from the wild-type allele to prevent the onset of tumorigenesis. Disease progression is contingent on the loss of the heterozygous state through inactivation of the wild-type allele, for example through deletion, which completely ablates expression of the tumor-suppressor activity. Subsequent research provided the molecular basis for the Knudson model, namely the necessity of homozygous inactivation of the retinoblastoma protein (Rb) on chromosome 13 for development of the disease (see Secs. 7.2.5 and 7.6.4).
7.2.4 Discovery of Oncogenes
In 1976, Michael Bishop and Harold Varmus isolated the oncogenic region of the Rous sarcoma virus, containing the v-src gene, by comparing the genetic content of a transformation-defective version of the virus with that of its highly transforming counterpart (Stehelin et al, 1976). Remarkably, they discovered that the normal avian DNA contained a nearly identical version of the v-src gene. On this basis, they advanced what was then a provocative concept, that oncogenes were altered versions of normal cellular genes, which they termed protooncogenes. Moreover, their findings suggested that other oncogenes might be found in the genomes of other transforming retroviruses. Indeed, many of the best known oncogenes were originally identified in viruses (see Chap. 6).
In 1982, the first somatically mutated cellular oncogenes were isolated in a number of laboratories by employing strategies that generally involved introducing DNA isolated from human cancer cell lines into mouse fibroblasts, which were then monitored for transformation. Isolation of individual genes from the transformed fibroblasts led to the identification of the activated mutant versions of the H-RAS and K-RAS oncogenes, derived from human bladder and lung carcinoma cell lines, respectively (Der et al, 1982). A year later, mutant N-RAS was similarly isolated from a neuroblastoma cell line (Shimizu et al, 1983).
7.2.5 Isolation of the First Tumor Suppressor
The prevailing view in the 1970s was that neoplastic transformation resulted from dominant activating mutations in oncogenes. This paradigm was challenged in the 1980s, when the retinoblastoma (Rb1) locus was mapped to a chromosomal region containing homozygous deletions, in both inherited and sporadic retinoblastomas (Cavenee et al, 1983). In 1986, a complementary DNA (cDNA) fragment mapping to the Rb1 locus was isolated and found to be at least partially deleted in retinoblastomas (Friend et al, 1986). The following year, 2 groups cloned the Rb cDNA, and reported that the transcript was either expressed in a truncated form or was entirely undetectable in tumors (Fung et al, 1987; Lee et al, 1987; Kallioniemi, 2008). Collectively, these results demonstrated the existence of a recessively acting cancer gene, with the homozygous loss of the wild-type Rb1 locus correlating with retinoblastoma development, as predicted by Knudson’s theoretical work 15 years earlier (see also Sec. 7.6.4).
7.2.6 The Genomics Age and Beyond
By the beginning of the 1990s, a clearer picture of the genetic changes accompanying cancer progression had begun to emerge, leading to the realization that cancer is a disease involving a combination of either inherited or somatic mutations, resulting in the activation of oncogenes or inactivation of tumor-suppressor genes (Fig. 7–2). Since then, additional putative oncogenes and tumor suppressors have been cataloged using both classical methods, as well as novel high-throughput analyses (Table 7–1; see Chap. 2, Sec. 2.2 for techniques). Functional characterization of proteins encoded by oncogenes and tumor-suppressor genes has led to an improved understanding of the processes responsible for tumorigenesis. It is now expected that a true understanding of cancer will require surveying for disease-associated changes beyond the genomic level. Of particular interest are epigenetic and microRNA factors, which influence normal cellular physiology (see Chap. 2, Sec. 2.4.3) and are often functionally altered during oncogenic progression.
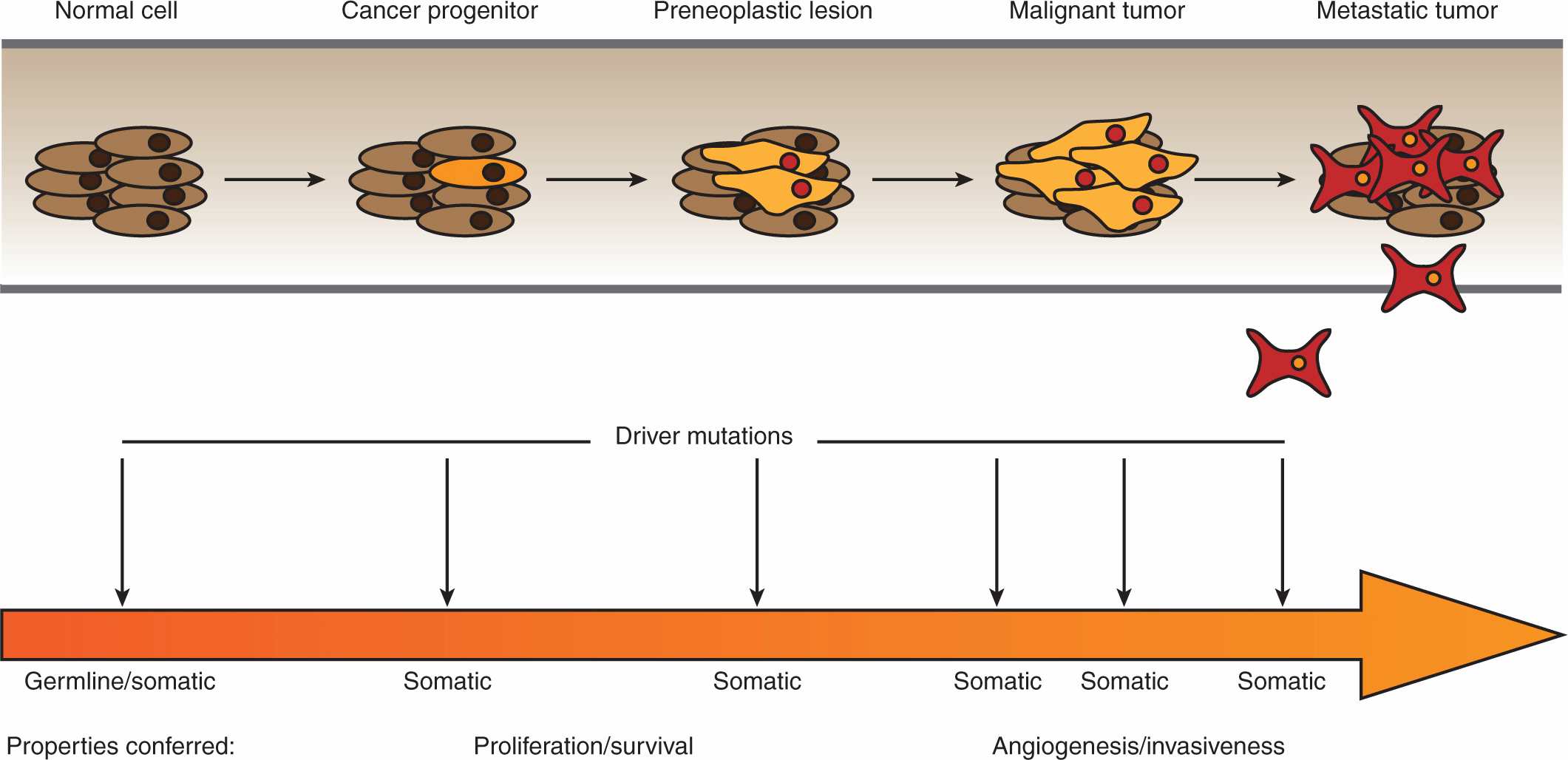
FIGURE 7–2 Multistage oncogenesis. During oncogenesis, a tumor progressively acquires a set of abnormal properties that enable it to support the growing mass of cells and ultimately metastasize to distal sites. It has long been recognized that this constitutes a multistep process involving a series of oncogenic hits, potentially occurring at both the germline (inherited) or somatic level. More recently, it has been appreciated that, in addition to genetic mutations, these hits can entail changes at the epigenetic, transcriptomic, proteomic, and metabolic levels. Moreover, these tumorigenic programs can be driven by different sets of oncogenic hits and progress at different rates.
TABLE 7–1 Oncogenes and tumor-suppressor genes.
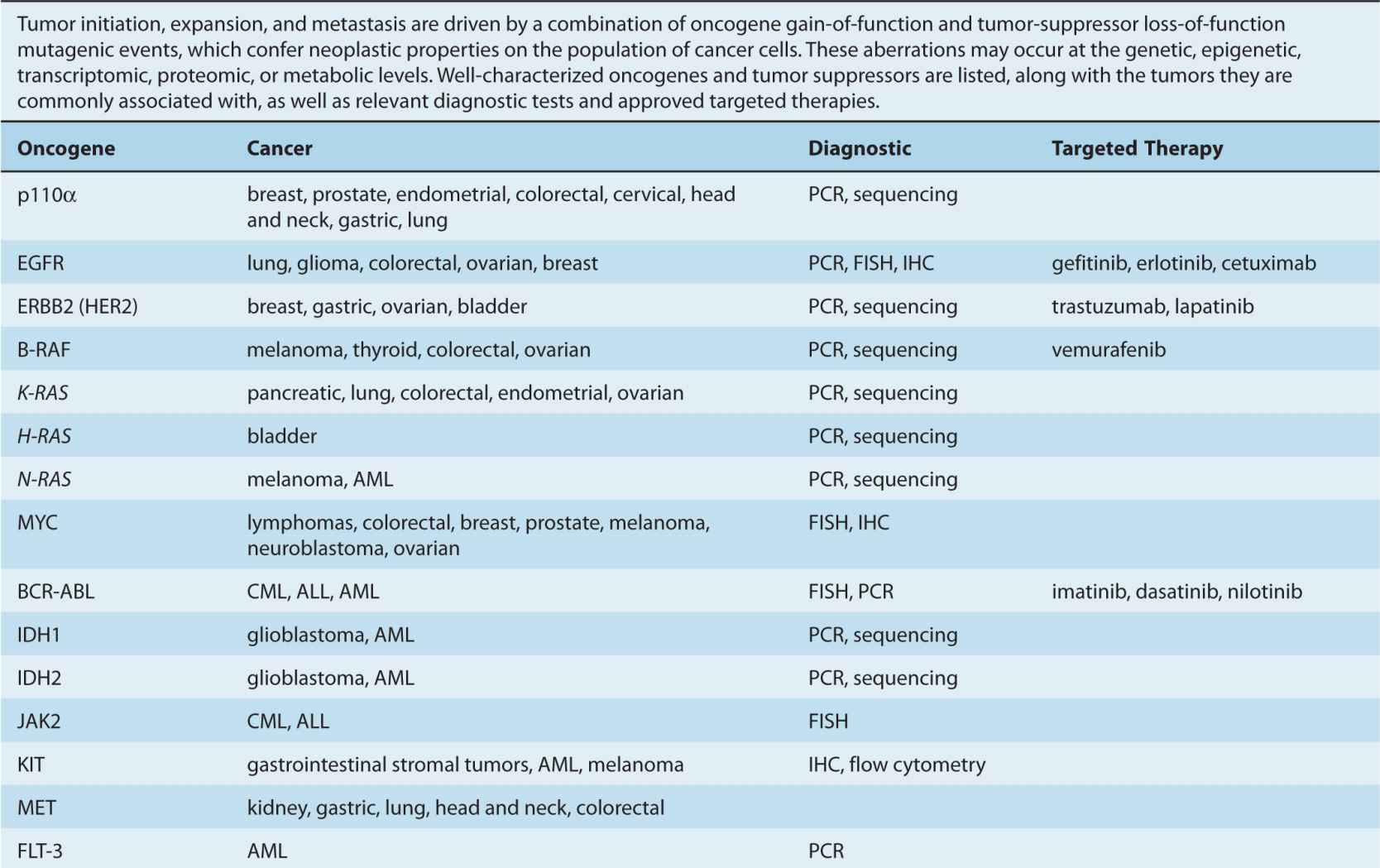
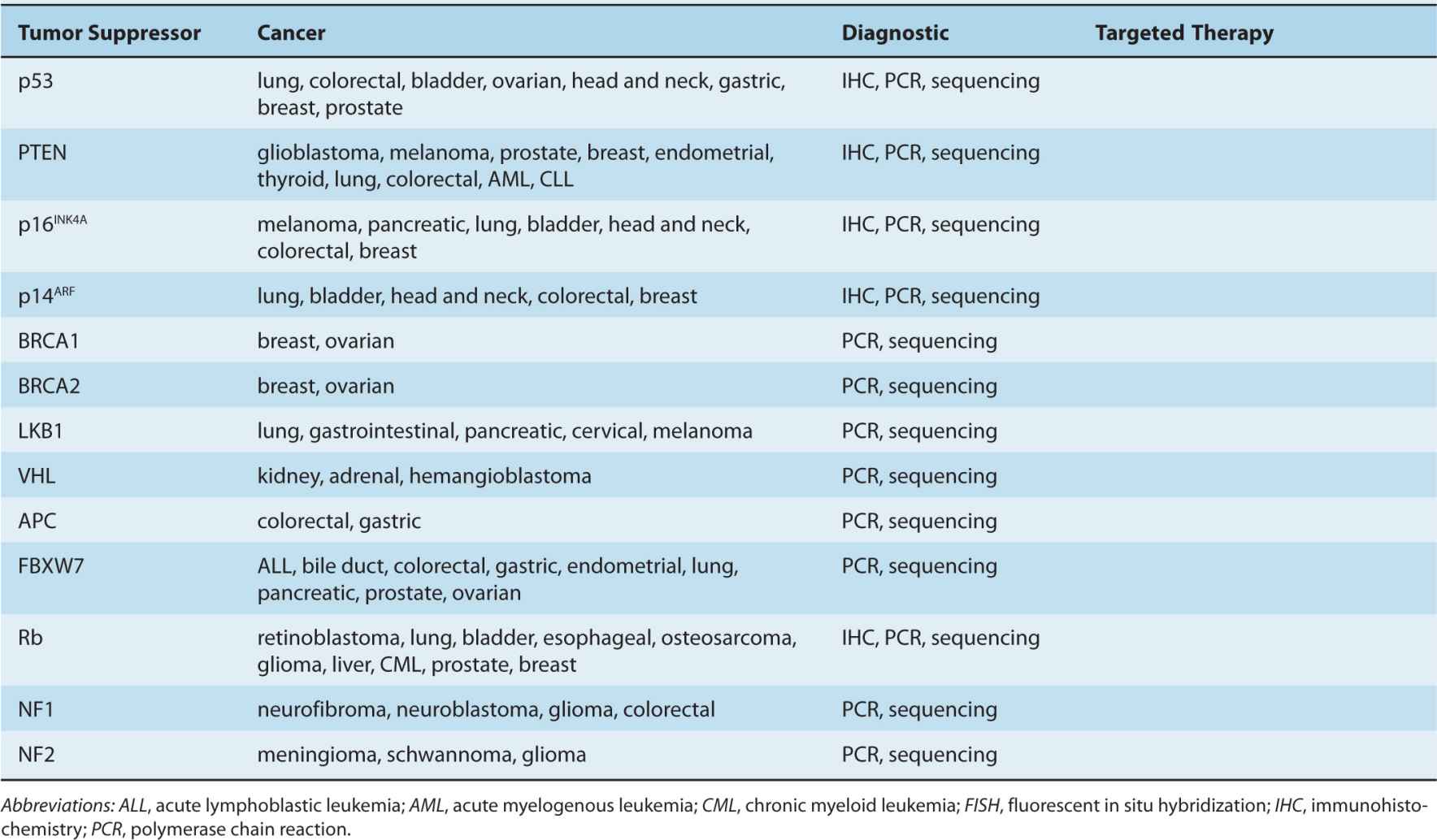
Comprehensive analyses of genomic, epigenetic, transcriptomic, proteomic, and metabolic states have been rendered practical for the first time by the advent of new high-throughput technologies (see Chap. 2). Characterizations of genomic structure including single nucleotide polymorphisms (SNPs), copy number variations, insertions, deletions, and translocations can be evaluated by SNP and comparative genomic hybridization (CGH) arrays, as well as next-generation sequencing (NGS) techniques (Carter, 2007; Kallioniemi, 2008; Ansorge, 2009). Epigenetic structure is being examined using chromatin immunoprecipitation (ChIP)-based NGS and array methods, while expression profiling, of both protein- and microRNA-encoding transcripts, is undertaken with both genome-wide and more specialized array platforms (Liu et al, 2008; Laird, 2010; see Chap. 2 for more details on these technologies).
There is great anticipation for insights that may be gleaned from these inventories of oncogenic changes at various levels of cellular regulation. Basic research scientists hope to develop a clearer picture of the underlying molecular mechanisms involved in oncogenesis by delineating the precise cellular events occurring at each stage of cancer progression. The prospects for translational scientists and clinicians are no less promising, encompassing the classification of cancers into more homogeneous subtypes, the identification of novel drug targets, and the development of prognostic tools capable of predicting metastatic propensity, therapeutic response, and survival (Fig. 7–3).
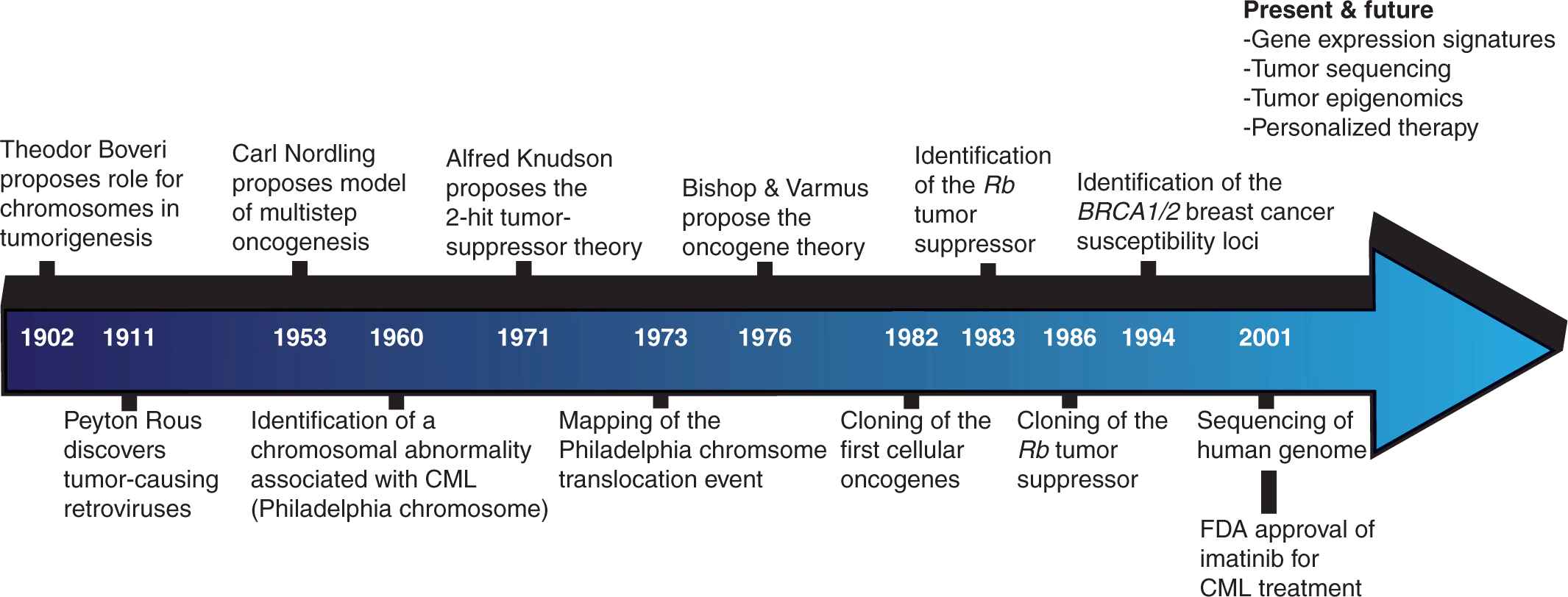
FIGURE 7–3 Timeline of cancer discoveries. For much of the past 2 millennia, our understanding of cancer remained relatively static. Beginning in the middle of the 19th century, technological improvements revolutionized our capacity to study the disease, and led to a number of paradigm-shifting discoveries. This process has accelerated in recent decades and we may now anticipate an era in which individual tumors can be comprehensively analyzed and personalized therapies designed to target the appropriate oncogenic mechanisms.
7.3 THE PROPERTIES OF NEOPLASTIC CELLS
The various tissues of the human body have evolved tightly regulated homeostatic mechanisms and lineage differentiation pathways that govern their function and regeneration if injured. Tumorigenesis is initiated when small populations of cells acquire genetic mutations that enable them to circumvent the homeostatic programs particular to their tissue microenvironment. The subsequent survival and expansion of the tumor mass is contingent on the acquisition of additional neoplastic properties, such as invasion. Ultimately, tumor cells gain metastatic potential, involving the capacity to migrate from their tissue of origin and invade distal sites to establish secondary malignancies (see Chap. 10).
Hanahan and Weinberg proposed that virtually all cancer cells exhibit certain hallmark properties: self-sufficiency in growth signals, insensitivity to antigrowth signals, evasion of apoptosis, limitless replicative potential, sustained angiogenesis, genomic instability, deregulated metabolism, the capacity for invasion and metastasis, as well as the ability to circumvent immune clearance an stimulate tumor-promoting inflammation (Hanahan and Weinberg, 2000; Hanahan and Weinberg, 2011; Fig. 7–4). Although a disproportionate number of cancer-associated mutations are found in a small group of critical regulatory proteins, each of these characteristics can be acquired through distinct sets of mutations, and the extreme genetic heterogeneity of cancer cells serves as a platform for the selection of tumor-promoting properties. Aberrant autocrine growth signaling, as well as hyperactivation of transmembrane receptors or intracellular signal transducers can engender self-sufficiency in growth signals. Acquired insensitivity to antigrowth signals often involves inactivation of the Rb protein or inhibition of terminal differentiation. Resistance to apoptotic death is frequently associated with loss of the p53 tumor suppressor, as well as the upregulation of antiapoptotic proteins such as BCL2. The replicative potential of normal cells is limited by the progressive attrition of telomeres at chromosome ends, a tendency counteracted in malignant cells through activation of the telomerase enzyme. The nutrient requirements of expanding tumors necessitates the de novo formation of blood vessels, triggered by a combination of upregulated proangiogenic factors, such as vascular endothelial growth factor (VEGF), and downregulation of inhibitory factors like thrombospondin-1 (see Chap. 11, Sec. 11.4). The spread of tumor cells to secondary sites requires the disruption of cell–cell and cell-extracellular matrix (ECM) contacts, as well as the capacity to penetrate tissue compartments, often through upregulation of extracellular protease activity (see Chap. 10).
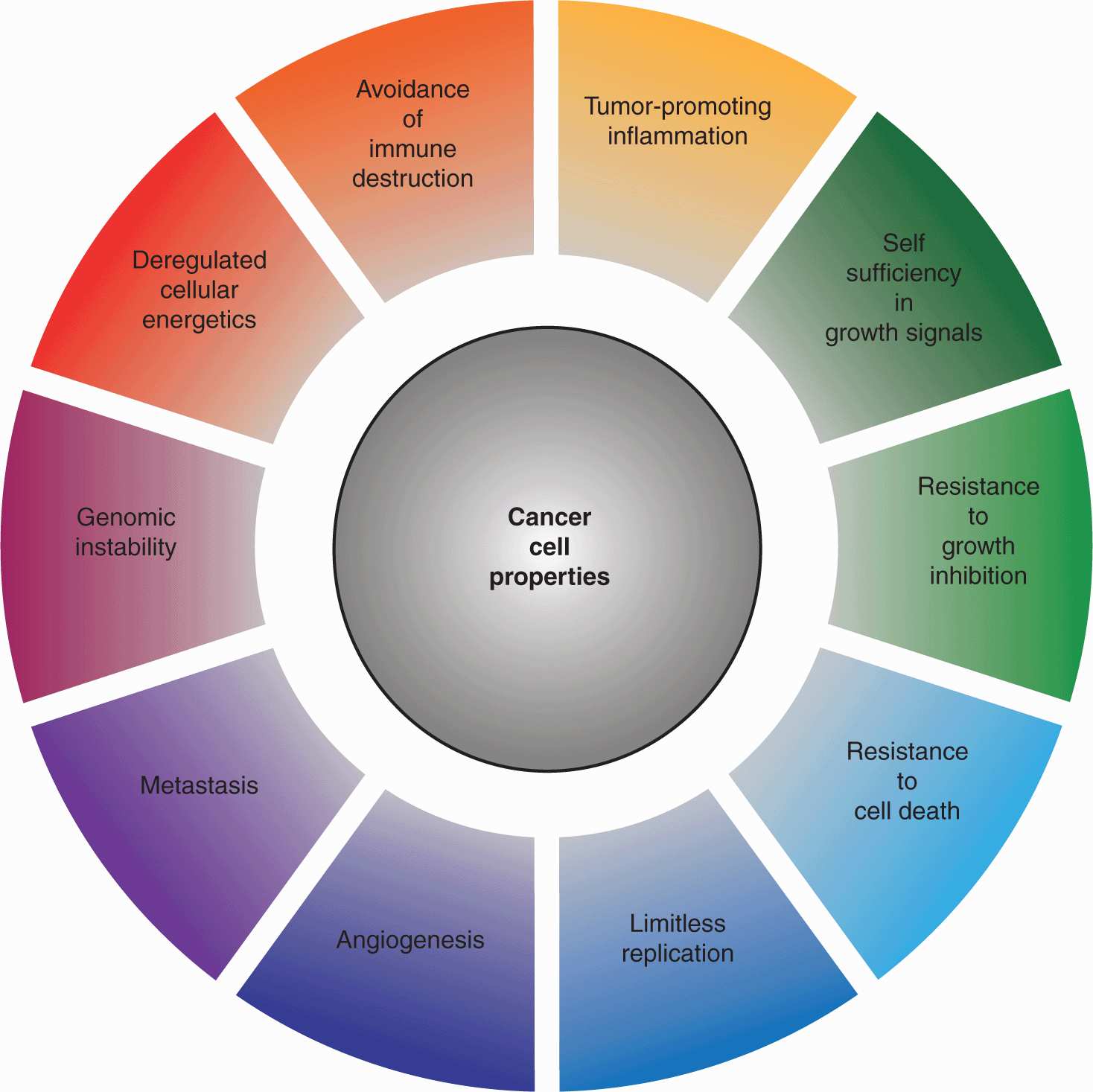
FIGURE 7–4 Properties of cancer. In 2000, Douglas Hanahan and Robert Weinberg proposed a model outlining a series of properties that distinguish cancer cells from their untransformed counterparts. In 2011, this model was updated to incorporate the findings of the ensuing decade of research. Collectively, these properties impart a capacity for tumor survival, expansion, and metastasis in circumstances in which growth would ordinarily be constrained by a variety of regulatory signals. (Adapted from Hanahan and Weinberg, 2011.)
Although the reductionist approach of characterizing tumors by these autonomous molecular changes in their cells can be beneficial, it is also important to conceptualize tumors within their tissue contexts. Solid tumors, in particular, are heterotypic collections of cells that include stromal compartments supporting the growth of the malignant cells. This idea was succinctly included in a model of oncogenesis advocated by Kinzler and Vogelstein, who classified cancer-associated genetic changes as gatekeeper, caretaker, and landscaper mutations (Kinzler and Vogelstein, 1996; Kinzler and Vogelstein, 1998; Fig. 7–5). The gatekeepers are generally tumor suppressors that normally constrain growth by regulating cell-cycle progression and activating apoptotic mechanisms as required. The caretakers, often DNA repair proteins, impede oncogenesis by preventing the genetic instability that favors the creation of tumorigenic mutations (see Chap. 5, Sec. 5.3). Alterations in stromal cells, which promote tumorigenesis were termed landscaper mutations.
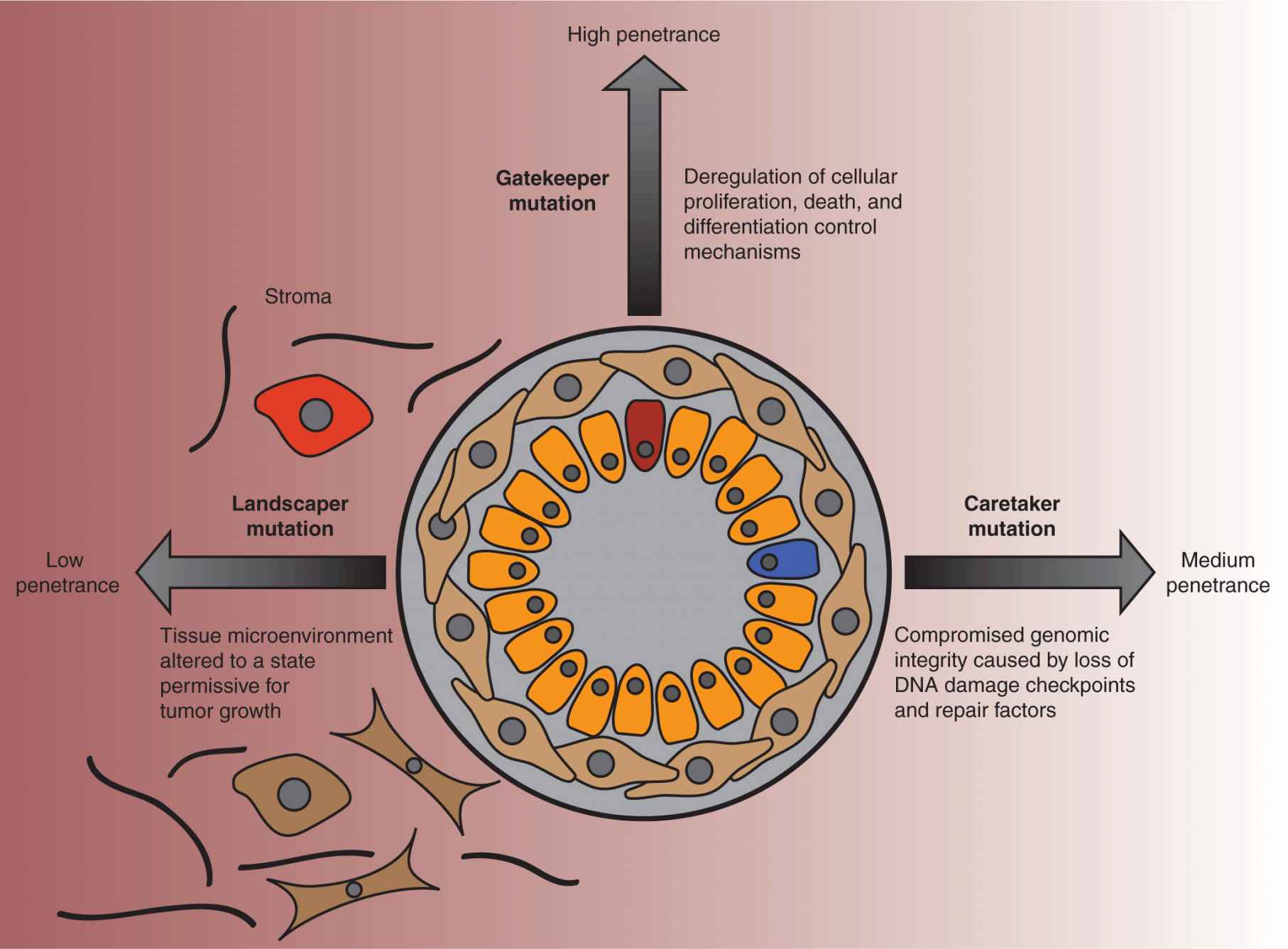
FIGURE 7–5 Gatekeeper, caretaker, and landscaper mutations. In the late 1990s, Kenneth Kinzler and Bert Vogelstein proposed a model of tumorigenesis based on their observations of the oncogenic mechanisms underlying hereditary colorectal cancer. Tumor-promoting genes were classified as gatekeepers, caretakers, and landscapers, according to their function. Gatekeeper genes encode proteins that regulate cell fate, be it proliferation, growth, arrest, death, or differentiation. Because deregulation of these mechanisms is likely to lead directly to abnormal growth, mutations in these genes correlate with the highest risk of cancer. Caretaker genes encode proteins that are responsible for guarding the integrity of the genome, with loss of function increasing the risk of acquiring mutations, for instance, in gatekeeper genes, that would lead to oncogenic growth. Mutations occurring in cells of the surrounding tissue stroma can create a microenvironment that is more permissive for tumorigenesis, and the associated genes were categorized as landscapers. As the functions of these genes are primarily supportive, in isolation, these mutations are much less penetrant.
Because genetic instability is a prominent feature of cancer cells (see Chap. 5, Sec. 5.2), the identification of proper therapeutic targets is dependent on determining which alterations represent causal driver mutations that promote oncogenic growth and which are neutral passenger mutations. Although many strategies are being pursued concurrently, there is as yet no clear method for distinguishing driver and passenger mutations (Carter et al, 2009; Bozic et al, 2010). Computational prediction methods have sought to identify driver mutations based on frequency of occurrence. Although it is tempting to categorize those alterations that occur at the background mutational rate as passenger mutations, rare mutations have sometimes been found to be potently oncogenic. Conversely, some genes located in mutational hot spots are disproportionately altered despite making no apparent contribution to oncogenic progression. Moreover, variations in background mutation rates between tumors complicate such analyses. It may be that as larger datasets become available, driver mutations will be found to occur at rates and in patterns that can be distinguished from passenger mutations, although this remains speculative. The presence of clusters of mutations, both within individual genes and known signaling pathways, is strongly suggestive of driver mutation status. Other prediction strategies aim to identify genetic changes that are likely to alter the function(s) of the encoded proteins. This is most obvious in the case of frameshift and nonsense mutations that introduce stop codons, or mutations that interfere with splice sites. Anticipating the effects of missense mutations, resulting in single amino acid changes, on the activity of a given protein is more challenging, but can be facilitated by the consideration of additional biological information, such as structural data.
7.4 THE BASIS OF TUMORIGENESIS
While cancer cells are defined at the cellular level by a handful of hallmark properties (see Fig. 7–4), at the molecular level, the genotypic and phenotypic changes are numerous, diverse, and complex. The genetic mutations that are responsible for cancer initiation are accompanied by additional alterations at the epigenetic, transcript, and protein levels (Fig. 7–6). Indeed, deregulation at any one of these levels may induce a cascade of additional mutagenic effects, creating the molecular basis for tumor expansion through clonal selection. In addition to point mutations, which can result in changes to amino acid sequences, other mutagenic events include gene amplifications, deletions, and insertions, as well as chromosomal translocations. The expression levels of cancer-associated genes can also be modulated epigenetically through promoter methylation, as well as the methylation, acetylation, and phosphorylation of histones (see Chap. 2, Sec. 2.3). Additional changes in the expression levels of proteins arise as a result of the modulation of transcriptional rates and messenger RNA (mRNA) stability, often involving the actions of small noncoding RNAs (see Chap. 2, Sec. 2.4.3). Much of the research effort has focused on the protein coding regions of the genome, the so-called exome, which represents only 1% to 2% of the entire human genome. The preponderance of aberrations occur in the remainder of the genome and their effects on oncogenesis, are as yet unclear. Ultimately, the deregulation of oncogenic targets, through an array of mutagenic mechanisms, is merely the means through which tumorigenesis confers the hallmark properties of cancer on transformed cells.
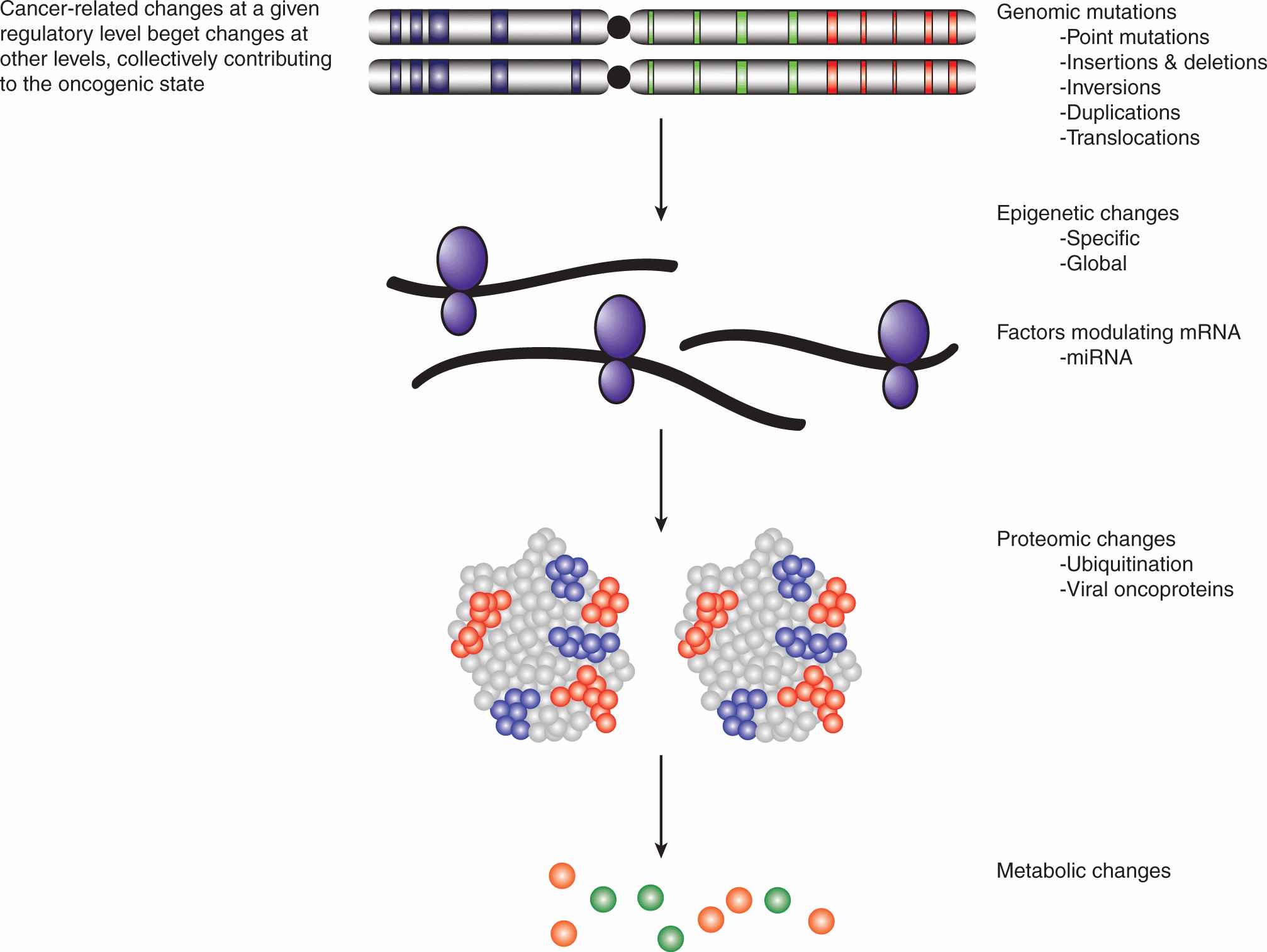
FIGURE 7–6 Cancer targets multiple regulatory levels. Cancer has long been recognized as a genetic disease, and large chromosomal abnormalities along with much smaller mutations have been cataloged since the 1960s. Previously, these were assumed to affect the function or expression of 1 or 2 proteins. More recently, broader changes affecting the epigenome, transcriptome, proteome, and the metabolic status of cells also have been associated with tumorigenesis. New oncogenic actors, such as microRNAs and oncometabolites, continue to emerge and contribute to a much more textured picture of the transformed phenotype. Moreover, oncogenic hits at any of these regulatory levels tend to engender further hits at other levels, synergistically promoting carcinogenesis. mRNA, Messenger RNA; miRNA, microRNA.
Specific oncogenes and tumor-suppressor genes are generally found to be mutated in some types of cancers but not others, and this varies among tumor subtypes that arise within the same tissue. This is puzzling when the gene in question is thought to influence fundamental cellular functions. Understanding why certain mutations or combinations of mutations occur preferentially in specific tumors is of fundamental importance, but remains speculative. The particular mutations capable of conferring neoplastic properties are dictated by the specific regulatory mechanisms that normally control these processes in the given tumor cell progenitor. Most cellular functions are regulated by multiple, partially redundant, regulatory networks. Because these networks have varying influence in different cell types, the severity of the effects caused by their disruption will also vary, and mutations in specific cancer genes will be subject to different selective pressures, depending on the cellular context in which they occur. Mutations that confer a selective advantage in one cell type may have little effect or even compromise the viability of another cell type. Factors likely to govern the effect of a particular mutation include the expression level of the gene, the existence of compensatory mechanisms, and developmental stage, as well as the modifying effects of other mutations.
7.4.1 Changes at the Genetic Level
The human genome is subject to a wide array of mutations of greatly differing sizes, ranging from point mutations affecting single nucleotides to large-scale rearrangements involving megabases of DNA (Fig. 7–7). Thousands of nucleotide variations have been cataloged in genome sequencing efforts, with many predicted to cause changes in the amino acid sequences of the encoded proteins, sometimes resulting in hyperactivation of oncoproteins or inactivation of tumor suppressors.
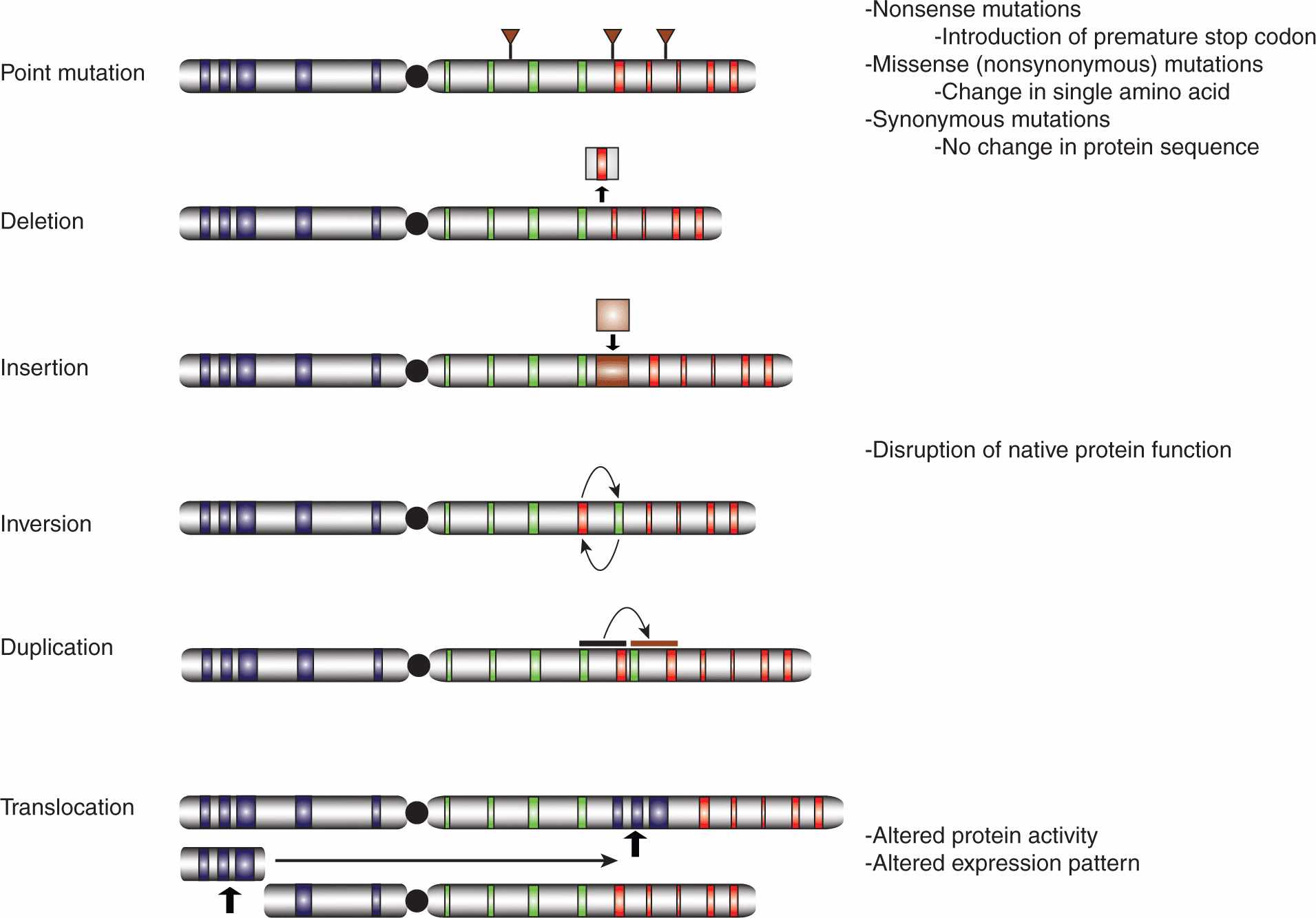
FIGURE 7–7 Classes of genetic mutations. The role of chromosome abnormalities in oncogenesis has been suspected for more than a century and specific genetic mutations have been identified since the 1960s. Whereas the available technologies once severely limited our capacity to characterize mutations, with next generation sequencing (NGS, see Chap. 2, Sec. 2.2.10), it is possible to characterize the entire set of genetic abnormalities in a given tumor cell in a relatively cost-effective manner. The number of genetic mutations in a given cancer cell is far greater than once suspected, encompassing point mutations affecting single nucleotides, deletions, insertions, inversions, and duplications, as well as intra- and interchromosomal translocations, all of varying sizes. Some mutations are functionally inert, while others can dramatically alter the activity and/or expression pattern of the affected protein(s).
Although gene amplifications and deletions have long been recognized as oncogenic events, the prevalence of such cancer-associated rearrangements in the human genome has probably been underappreciated. Since the discovery of the translocation event resulting in the generation of the oncogenic BCR-ABL fusion gene, additional cancer-associated gene fusions have been identified, particularly in hematological cancers, as well as in bone and soft-tissue sarcomas (Nussenzweig and Nussenzweig, 2010). Increasingly, such genomic rearrangements are also being identified in epithelial-derived malignancies, including prostate, lung, and breast tumors (Table 7–2) (Kumar-Sinha et al, 2008; Edwards, 2010).
TABLE 7–2 Cancer-associated gene fusions.
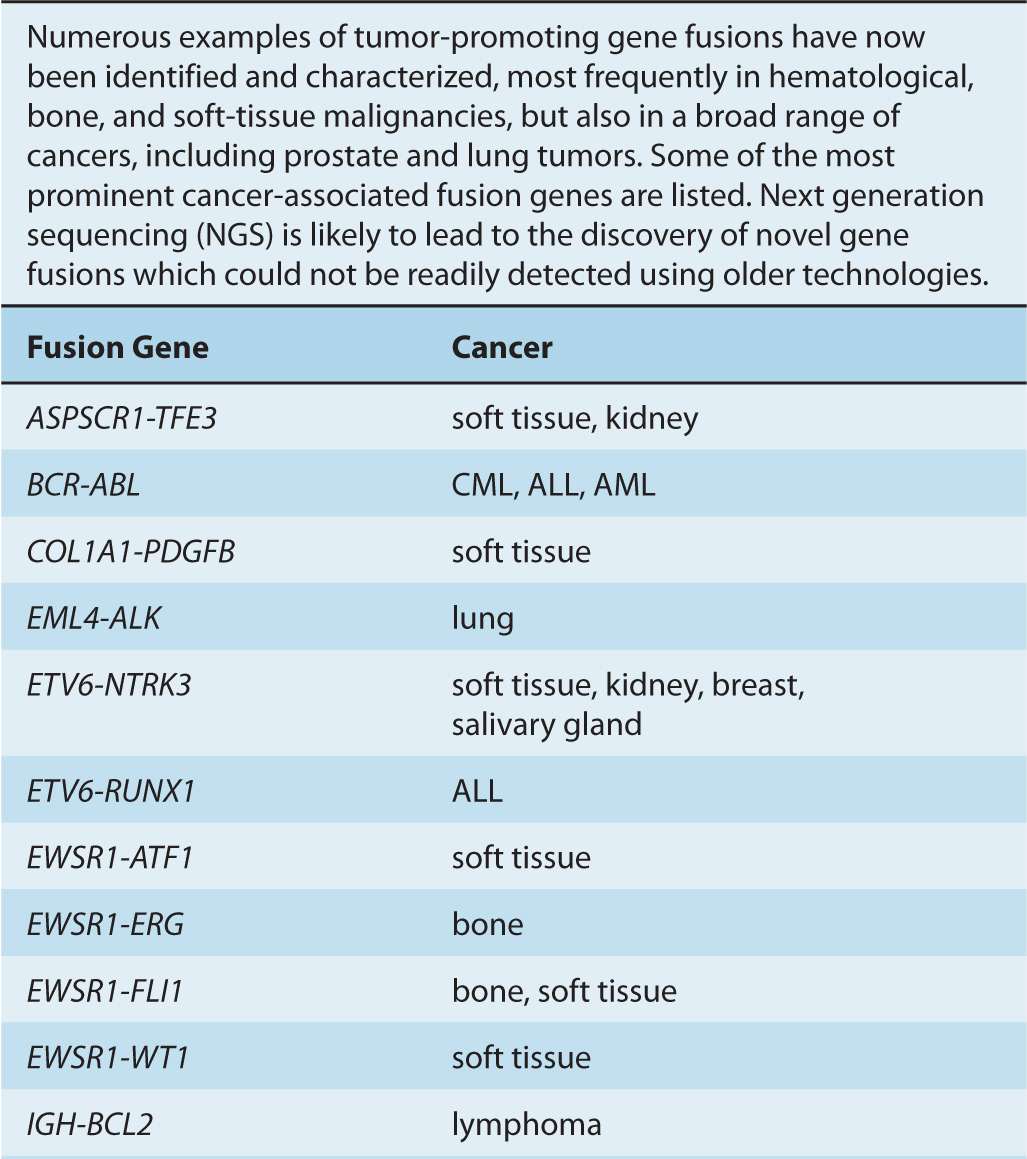
Gene fusions result from intra- or interchromosomal translocations, as well as deletions, inversions, and tandem duplications (see Fig. 7–7). In some instances, the expression of one of the genes is altered as a result of its juxtaposition with the transcriptional regulatory elements of the second gene (see Fig. 7–8). Some follicular lymphomas feature translocation events between chromosomes 14 and 18 that bring the BCL2 gene, encoding the antiapoptotic BCL2 protein, under the control of the immunoglobulin (Ig) heavy-chain gene enhancer, rendering the cells resistant to cell death signals. Gene fusions that result in the addition or deletion of regulatory domains can cause constitutive activation of the affected protein, as a result of forced multimerization, differential sub-cellular localization, or altered protein-protein interactions. Recombination between chromosomes 9 and 12 is found in leukemic cells of CML and acute lymphoblastic leukemia (ALL), resulting in production of the constitutively active TEL-JAK fusion tyrosine kinases that regulate cell proliferation, survival, and differentiation.
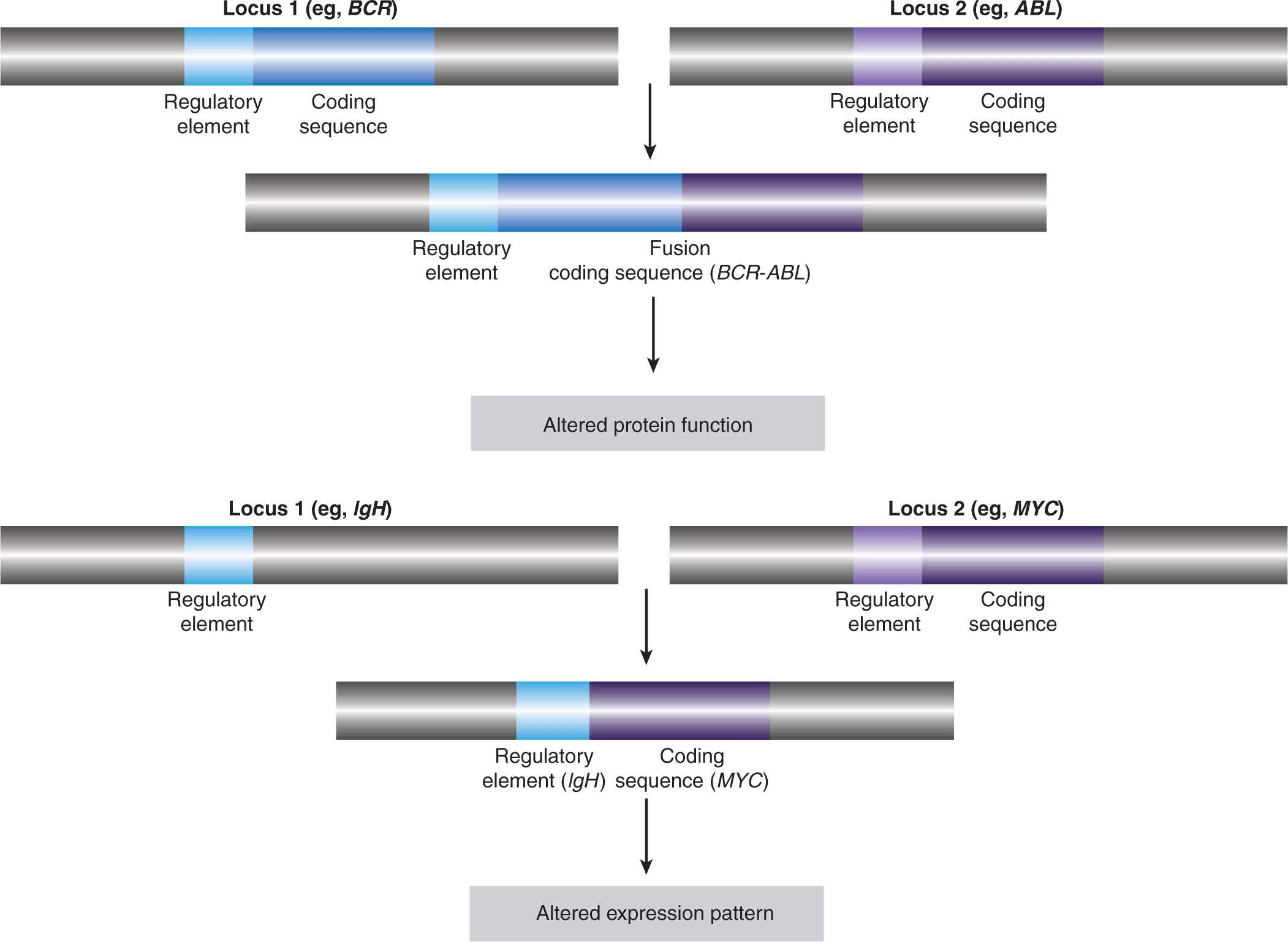
FIGURE 7–8 Effects of translocations. The first observed cancer-associated chromosomal abnormality was a reciprocal translocation between chromosomes 9 and 22, resulting in the so-called Philadelphia chromosome, identified in CML patients. The functional result of this genetic event is the creation of the BCR-ABL fusion protein, which causes the constitutive activation of the ABL kinase. This represents an example of a chromosomal translocation that alters the activity of the affected protein(s). In other translocations, it is the expression pattern of the protein that is altered, rather than its structure or direct function. One such example is the juxtaposition of the MYC gene with powerful transcriptional regulatory elements of the immunoglobulin heavy chain (IgH) observed, for instance, in Burkitt lymphoma, which results in a dramatic upregulation of the MYC transcription factor. Other examples are described in the text and in Table 7–2.
On the basis of analyses of cancer genomes, it is suspected that most epithelial carcinomas also contain gene fusion events. Notably, more than half of all prostate cancers harbor a TMPRSS2-ERG fusion event, caused by an intrachromosomal deletion on chromosome 21, in which the gene encoding the ERG transcription factor is brought under the control of the androgen-dependent regulatory elements of the TMPRSS2 gene promoter, leading to its aberrant expression (Clark and Cooper, 2009). A small percentage of non–small cell lung cancers feature a fusion between the EML4 gene and the ALK tyro-sine kinase gene, which results in forced oligomerization and constitutive activation of the kinase (Crystal and Snow, 2011).
Gene amplification events produce regions of DNA (amplicons) containing multiple copies of oncogenes, such as the breast cancer-associated ERBB2 (HER2) or MYC, and effectively achieve hyperactivation of oncogenic pathways by dramatically increasing oncogene dosage. The deletion of regulatory modules, such as the epidermal growth factor receptor (EGFR) ligand-binding domain, can also result in oncogenic deregulation of proteins. Conversely, the deletion of tumor-suppressor genes, such as PTEN, is also a common oncogenic event. Point mutations can affect enzymatic activity, regulatory phosphorylation sites, protein–protein interactions, protein stability, and subcellular localization. Moreover, many of the oncoproteins that are initially sensitive to a given therapeutic agent often acquire secondary mutations that render them refractory to the drug.
7.4.2 Changes at the Epigenetic Level
A number of heritable characteristics could not be solely attributed to genetics, and the field of epigenomics has sought to explore these factors (Fletcher and Houlston, 2010; see Chap. 2, Sec. 2.3). Epigenetic effects entail heritable changes in gene expression that are not the result of alterations in the primary DNA sequence and they can influence oncogenic progression. These events may also represent novel targets for therapeutic intervention (Fig. 7–9).
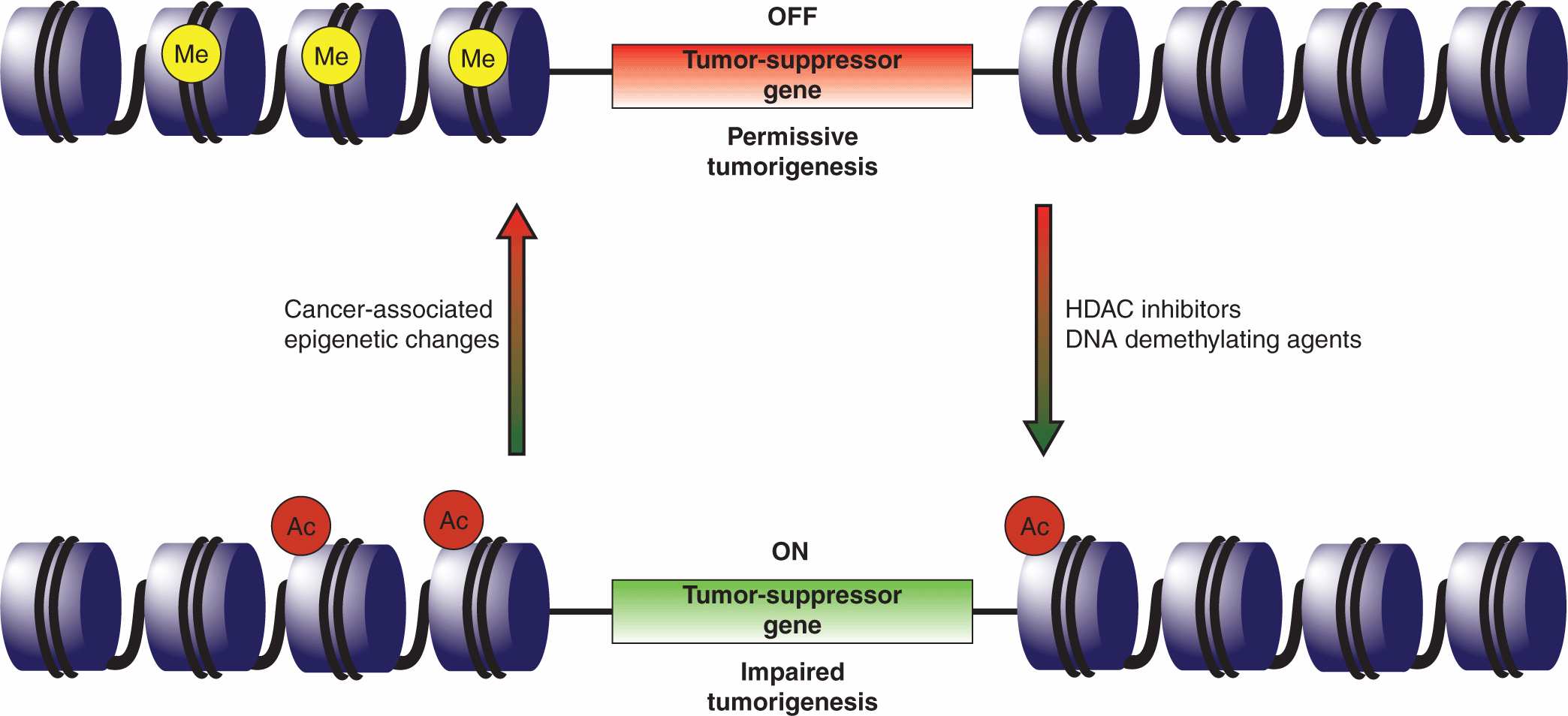
FIGURE 7–9 Epigenetic therapy. Global changes in the cellular epigenome frequently accompany transformation and the epigenetic silencing of tumor-suppressor genes or activation of oncogenes is now a recognized tumorigenic mechanism. One such example is the silencing of the p16INK4A and antigen-presenting cell (APC) tumor-suppressor genes in non–small cell lung cancers. Clinical testing suggests that the tumor-suppressor genes can be reactivated through the application of DNA methyltransferase inhibitors (eg, 5’-deoxycitidine), to reduce the methylation of promoter regions, and histone deacetylase (HDAC) inhibitors (eg, entinostat) that activate gene loci through chromatin remodeling. It is anticipated that epigenetic therapies will be most effective in a combinatorial setting, enhancing the response to other anticancer agents.
The preeminent epigenetic factors are DNA methylation and a pattern of acetylation, methylation, and phosphorylation of DNA-associated histone proteins that are collectively known as the histone code. The methylation pattern of the cellular genome, whereby DNA methyltransferases attach methyl groups to cytosines within CpG dinucleotides, specifies which genes are actively transcribed or silenced. The CpG dinucleotides are enriched within the so-called CpG islands that span the 5′ ends of some genes, typically including the promoter and first exon (Esteller, 2008). In mammals, the majority of genes have promoters containing CpG islands, and promoter methylation results in the silencing of the associated gene(s). Histone modifications alter localized chromatin structure, rendering regions either amenable or resistant to transcription. The various histone modifications do not occur independently of one another, but rather entail a complicated interplay that collectively controls gene expression.
Tumorigenesis frequently appears to coincide with both global and local epigenetic changes that are in some respects paradoxical (Chi et al, 2010; Veeck and Esteller, 2010). At a global level, the genome typically becomes hypomethylated during oncogenic progression. A sizable portion of the human genome consists of repetitive stretches of identical or similar sequences, a subset of which are transposable DNA elements that have the capacity to alter their genomic location. The methylation of these repetitive sequences is thought to block the activation and movement of transposable elements, and therefore prevent chromosomal instability. Thus, global hypomethylation of the genome may contribute to the genomic instability that is a prominent feature of transformed cells (see Chap. 5, Sec. 5.2.1).
Promoter methylation patterns of critical cancer-associated genes also feature prominently during tumorigenesis, with hypermethylation-associated silencing of tumor-suppressor genes and hypomethylation-mediated activation of onco-genes. Cancer-associated hypermethylation of the Rb1 tumor-suppressor gene promoter was noted in the 1980s, and similar epigenetic targeting of other tumor suppressor promoters, including those for BRCA1 and PTEN, has since been reported. A detailed understanding of the role of epigenetics in neoplastic transformation remains a work in progress. For instance, it is not clear whether the cancer-associated epigenetic modifications result from specifically targeted acts or whether they represent random epigenetic changes that are then selected for in the course of oncogenic clonal expansion.
7.4.3 Changes at the Transcript Level; microRNAs
One of the great revelations of recent biological research has been the discovery of the extensive roles of small RNAs in the control of gene expression. MicroRNAs are 20- to 30-nucleotide RNA molecules, generated by defined posttranscriptional maturation steps, that hybridize to complementary sequences in target mRNA transcripts in order to block gene expression (see Chap. 2, Sec. 2.4.3). This may be accomplished either by promoting transcript degradation or by interfering with the translation process. It is now recognized that microRNAs govern a variety of cellular processes, regulating at least one-third of all human genes, and may have key roles in tumorigenesis. MicroRNAs that normally suppress transformation by targeting oncogenic transcripts are downregulated in some cancers, whereas those targeting tumor-suppressor transcripts are sometimes overexpressed (Croce, 2009; Garzon et al, 2009; Ryan et al, 2010).
Genes encoding microRNAs, no less than proteinencoding genes, are subject to mutations and epigenetic silencing resulting in loss of function. MicroRNA expression can also be affected by oncogenic changes occurring at any of the post-transcriptional biogenesis steps whereby the immature RNA molecule is processed into the mature microRNA form. Deletions in the 13q14 chromosomal region are common abnormalities associated with chronic lymphocytic leukemia (CLL). The underlying molecular cause was unclear as no protein-coding tumor-suppressor gene could be identified in the region. The nature of the oncogenic mechanism was only clarified when the miR-15/miR-16 microRNA gene cluster was localized to this segment and found to target the antiapoptotic BCL2 protein, which is found at elevated levels in CLL samples (Cimmino et al, 2006). The LET-7 microRNA family members, which target the RAS and MYC onco-genes, are downregulated in many cancers, including lung, breast, and colon tumors (Johnson et al, 2005; Sampson et al, 2007). Intriguingly, miRNA-29 has been shown to target the DNMT3A methyltransferase and thus could affect onco-genesis through global epigenetic effects (Fabbri et al, 2007). MicroRNAs are potentially attractive therapeutic focal points because individual molecules regulate multiple targets, theoretically affecting several oncogenic pathways simultaneously. because individual molecules regulate multiple targets, theoretically affecting several oncogenic pathways simultaneously.
7.4.4 Changes at the Protein Level
Virally encoded oncoproteins often trigger tumorigenic programs by directly interacting with critical host cell regulatory proteins. The first identified human tumor virus, the Epstein-Barr herpesvirus was discovered in the 1960s when Joseph Epstein detected viral particles in Burkitt lymphoma biopsies (Caparros-Lefebvre et al, 1996; Klein et al, 2010). As most adults are Epstein-Barr virus (EBV) carriers without developing the disease, the oncogenic mechanisms are not certain, but virus-encoded proteins appear to be involved in a complex interplay with proteins of host B-cell lymphocytes, including the p53 and Rb tumor suppressors (see Chap. 6; Sec. 6.2.4).
Additional human tumor viruses have since been identified, with roughly 20% of all cancers thought to have at least some viral etiology (see also Chap. 6, Sec 6.2). The hepatitis C (HCV) RNA virus constitutes a major hepatocellular carcinoma risk factor. Although a comprehensive understanding of HCV-associated hepatocarcinogenesis remains elusive, it appears that virally encoded proteins affect host cell physiology without proviral integration (Tsai and Chung, 2010). Viral proteins disrupt several signaling pathways, again including checkpoint mechanisms activated by the p53 and Rb tumor suppressors. Sequences of the human papillomavirus (HPV) have been detected in human malignancies, notably cervical cancers (Munger et al, 2004) and oropharyngeal carcinomas (Miller et al, 2012). The HPV E6 gene product inactivates the p53 tumor suppressor protein and induces expression of the TERT subunit of the telomerase enzyme (see Chap. 5, Sec. 5.7). The E7 gene product destabilizes the Rb tumor suppressor, can override the inhibitory effects of the cyclin-dependent kinase (CDK) inhibitors p21CIP1 and p27KIP1, and interacts with a number of other host proteins, including his-tone deacetylases and acetyltransferases. Although there are a multitude of virus–host protein interactions, inactivating interactions between virus-encoded proteins and p53 or Rb are a recurring theme in viral–host pathogenic interactions (Levine, 2009). A detailed discussion of viruses in cancer can be found in Chapter 6.
7.5 GAIN OF FUNCTION EVENTS
7.5.1 BCR-ABL
The Philadelphia chromosome was initially described initially as a minute chromosome and assumed to be the product of a loss of genetic material from chromosome 22 (Nowell and Hungerford, 1960). Improvements in cytogenetics permitted more careful examination of chromosome defects, allowing Janet Rowley to determine that the deletion of genetic material in chromosome 22 was matched by a comparable insertion in chromosome 9 (Rowley, 1973). She speculated that the chromosomal defects observed in CML cases were the result of a translocation exchange between chromosomes 9 and 22. By 1985, it had been determined that cells of most patients with CML feature reciprocal translocation events between the long arms of the 2 chromosomes, resulting in the creation of a BCR-ABL hybrid gene (see Fig. 7–8). The same translocation event has been found to occur in 25% to 30% of adult and 2% to 10% of pediatric ALL, and occasionally in cases of acute myelogenous leukemia (AML) (De Klein et al, 1986).
The ABL kinase is the human homolog of the transforming sequence found in the Abelson murine leukemia virus. Whereas the protein normally shuttles between the nucleus and cytoplasm, the product of the BCR-ABL fusion gene is a constitutively active tyrosine kinase permanently localized in the cytoplasm. The addition of the BCR gene product appears to promote extensive multimerization of the fusion protein, facilitating self-activation by trans-autophosphorylation. During the chronic phase of CML, the BCR-ABL kinase activates a number of downstream pathways while also increasing genomic instability. In the absence of therapeutic intervention, additional genomic aberrations ultimately lead to a transition from the chronic to the advanced CML phase known as blast crisis that is more difficult to treat. At a cellular level, the blast phase is marked by increased proliferation accompanied by impaired apoptosis and differentiation, which together increase the population of blast progenitors. One noteworthy molecular change is a moderate increase in BCR-ABL levels, enhancing throughput of the relevant signaling pathways and begetting still more genetic damage.
The correlation of a disease state with a particular genetic lesion offered the possibility for targeted therapeutic intervention. The subsequent development and successful application of imatinib, which binds in the vicinity of the adenosine triphosphate (ATP)-binding site and locks BCR-ABL in an inhibited conformation, remains one of the most celebrated achievements in targeted cancer therapy.
7.5.2 MYC
MYC family members are global transcription factors (see Chap. 8, Sec. 8.2.6) that activate some target genes while repressing others, thereby affecting numerous cellular processes including proliferation, growth, apoptosis, and angio-genesis (Fig. 7–10) (Meyer and Penn, 2008; Albihn et al, 2010). Roughly 15% of all human genes are thought to be regulated by MYC proteins, so it is difficult to pinpoint the specific MYC targets that are responsible for oncogenesis. For example, MYC promotes cell-cycle progression by influencing a variety of targets, including downregulation of CDK inhibitors and upregulation of CYCLIN D1, CDK4, CDC25A, and E2F transcription factors (see Chap. 9, Sec. 9.2.2). Numerous genes, associated with the production of the constituent building blocks required for cell growth, are also controlled by the MYC proteins. Importantly, MYC deregulation appears to correlate with general chromosomal instability, although there is not yet agreement about the responsible mechanisms. Paradoxically, MYC upregulation also triggers apoptosis, and additional oncogenic mutations capable of blocking cell death are required for tumor progression. Thus, MYC overexpression results in pleiotropic downstream effects that collectively promote oncogenic transformation.
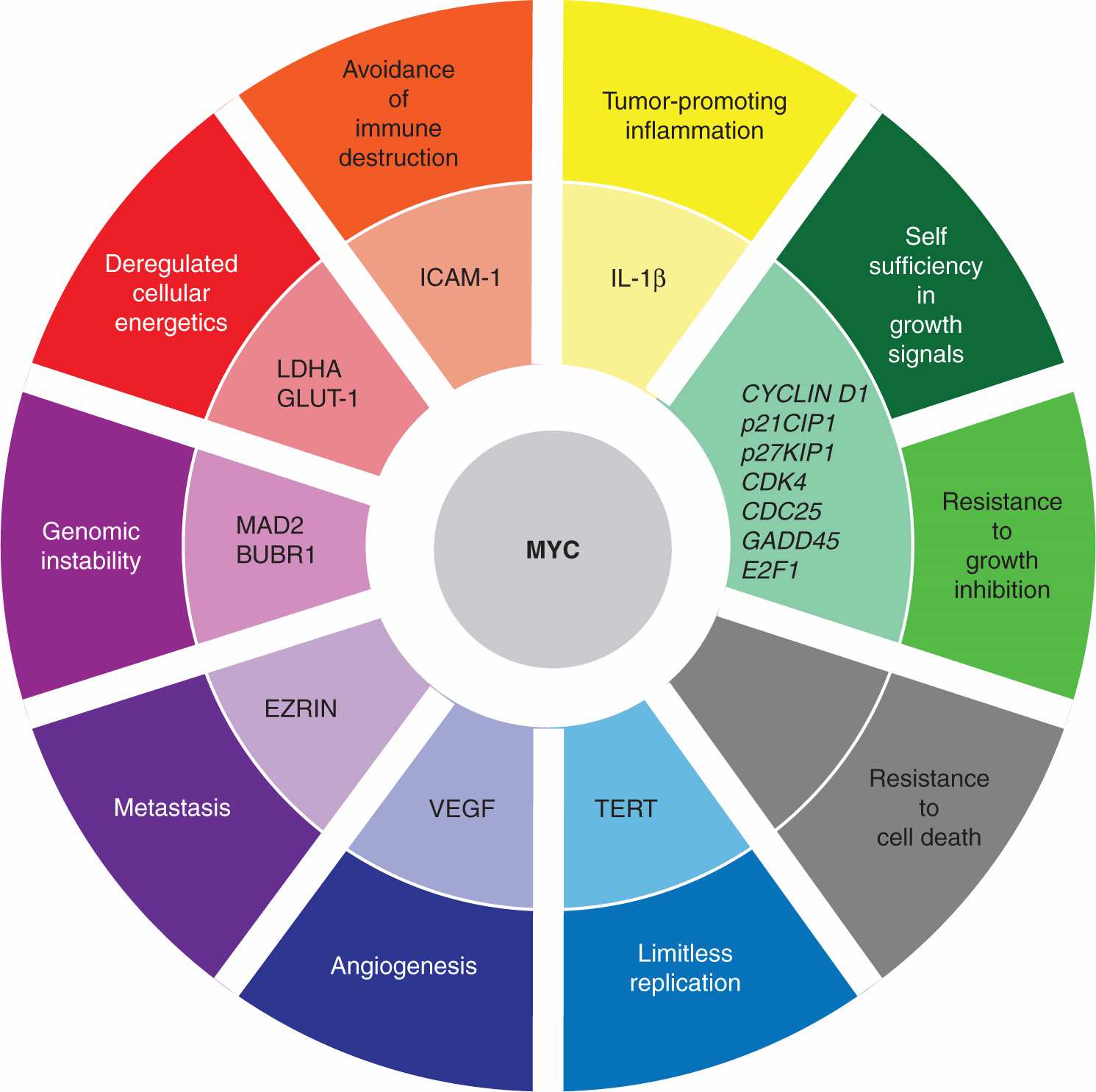
FIGURE 7–10 MYC targets and the promotion of tumorigenesis. The MYC transcription factor is thought to regulate the expression of roughly 15% of all human genes and is frequently upregulated in a variety of cancers. MYC upregulates the expression of some genes while repressing the expression of other targets. Unsurprisingly, given the number of genes affected by MYC, its deregulation contributes to the acquisition of many of the characteristic properties of cancer cells, as defined by Hanahan and Weinberg (2011). One notable exception is the paradoxical induction of apoptosis by MYC overexpression, necessitating the disengagement of cell death pathway(s) by additional oncogenic hits so as to foster transformation. Some of the MYC-regulated genes involved in tumorigenesis are denoted, although the particular MYC targets critical to oncogenesis will undoubtedly vary in different tumors.
Transforming viruses can direct oncogenesis by proviral DNA integration in the proximity of protooncogenes or tumor-suppressor genes, leading to altered expression or splicing (see Chap. 6, Sec. 6.3). An interesting example is the mechanism of chicken B-cell lymphogenesis by the slowly transforming avian leukosis virus (ALV). The ability of ALV to induce lymphomas was initially puzzling because of the apparent absence of any transforming sequence in the viral genome. Nevertheless, a careful analysis of lymphomas from independently infected birds revealed proviral integration at highly specific sites, giving rise to similar viral–host hybrid RNAs (Hayward et al, 1981). The viral coding segments were often altered in ways that precluded their transcription, indicating that viral protein expression was not required for transformation. Remarkably, the hybrid RNAs were found to include Myc coding sequences leading to increased expression of myc protein, suggesting that proviral integration promoted oncogenesis through upregulation of myc levels. Lymphomas in birds caused by ALV infection are slow growing as the oncogenic mechanism involves clonal selection of the rare proviral integration events in the vicinity of the myc protooncogene. This constituted the first observation of neo-plastic transformation caused by the upregulation of a non-mutated cellular gene. Interestingly, the acutely transforming avian myelocytomatosis retrovirus (MC29), from which MYC derives its name, induces leukemia in birds through expression of v-Gag-Myc already encoded in the viral genome (Reddy et al, 1983).
Upregulation of Myc expression is observed in some mouse plasmacytomas as a result of a recombination between the Myc and immunoglobin (Ig) heavy-chain genes. Burkitt lymphomas, as well as other human B-cell lymphomas, frequently feature chromosome translocation events between chromosome 8, where the MYC gene is located, and chromosomes 14, 2, or 22, where the Ig heavy- and light-chain genes reside. These rearrangements have the effect of bringing the MYC gene under the transcriptional control of Ig gene enhancer elements, leading to its overexpression. Whereas genomic translocations involving MYC genes are common in hematopoietic cancers, heightened MYC levels are often attained in solid tumors through gene amplification. The MYCN gene, which is normally expressed during development, has been found amplified in neuroblastomas, and increased copy numbers of the related MYCL1 gene have been observed in several cancers, including ovarian tumors.
7.5.3 EGFR/ERBB2
The involvement of epidermal growth factor (EGF) signaling in tumorigenesis has been recognized for 3 decades. The EGF family is comprised of 11 related ligands that stimulate intra-cellular signaling by binding to the EGF receptors, ERBB1 (EGFR), ERBB3, and ERBB4 (Baselga and Swain, 2009). A fourth member of the EGF receptor family, ERBB2 (HER2), cannot bind ligands itself, but is activated by dimerization with other family members (see also Chap. 8, Sec. 8.2.1). The EGF receptors are transmembrane proteins consisting of an N-terminal ligand-binding ectodomain, an intracellular kinase domain, and a C-terminal regulatory tail that contains docking sites for a number of downstream effectors. Ligand binding to the ectodomain induces receptor dimerization, transmitting conformational changes that promote kinase activation. Active receptor signaling complexes can be formed by both homodimerization and certain heterodimer combinations. The ERBB3 receptor binds ligands but has an inactive kinase domain and therefore can only transmit the EGF signal as part of a heterodimer with an active family member (Fig. 7–11).
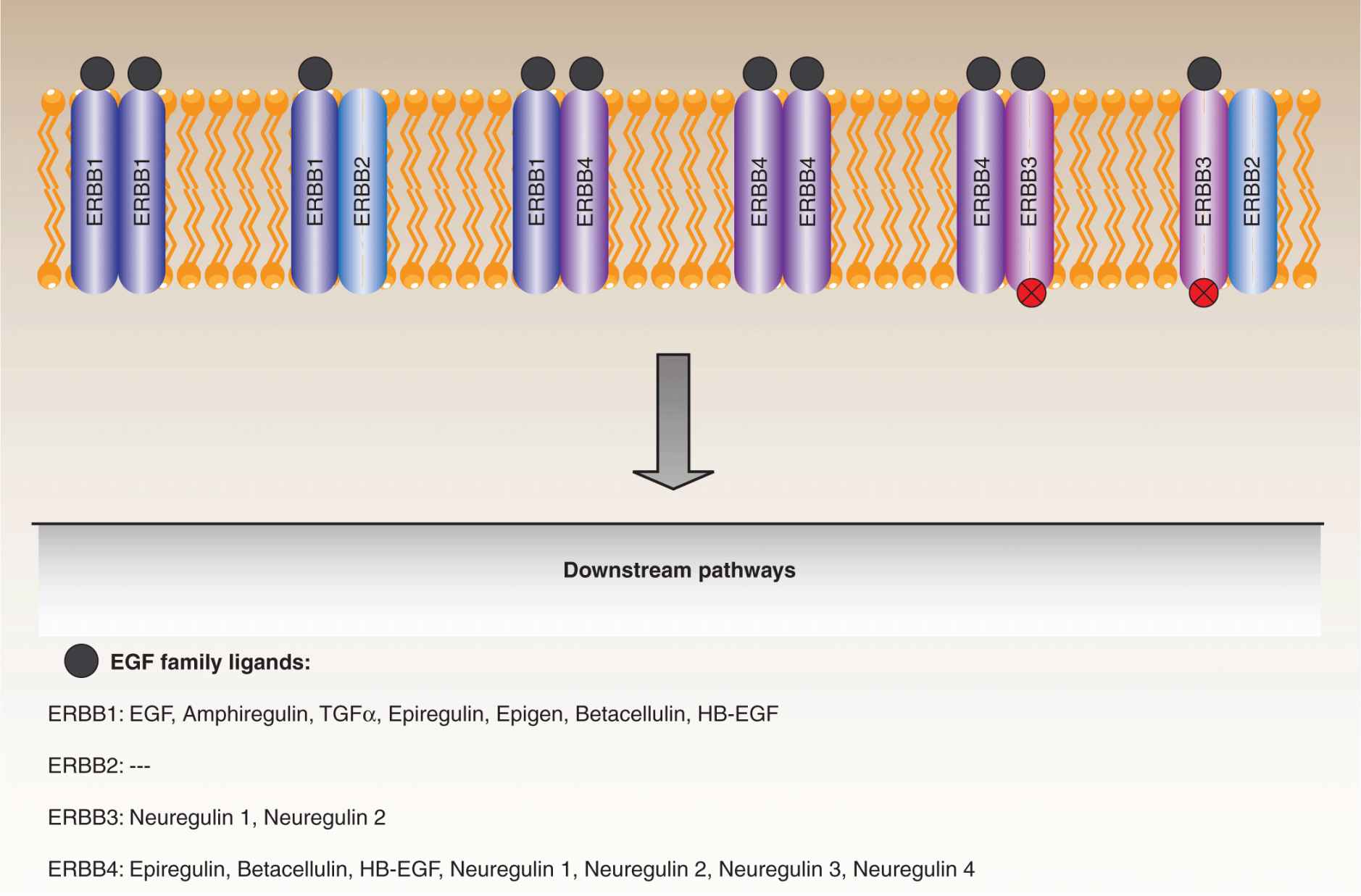
FIGURE 7–11 EGF receptor family. The EGF receptor family consists of 4 transmembrane proteins that couple a ligand-binding domain that responds to external stimuli with intracellular tyrosine kinase domains responsible for signal transduction. ERBB1 (EGFR), ERBB3, and ERBB4 each respond to a particular set of EGF family ligands, with some overlap, whereas ERBB2 appears to be an orphan receptor that must dimerize with ERBB1 or ERBB3 in order to engage downstream pathways. In addition, ERBB3 has an inactive kinase domain and transmits signals by dimerizing with ERBB2 or ERBB4. Collectively, these receptors regulate pathways that impact a number of cell processes, and are deregulated in a number of cancers.
All 3 portions of the ERBB1 receptor are subject to oncogenic mutations (Pines et al, 2010). Ligand-independent dimerization occurs as a result of point mutations or, more commonly, deletions within the ectodomain that leave the receptors in a constitutively activated conformation. Many such deletion mutants have been characterized, with one notable example being the variant III mutant, lacking the exons 2 to 7 coding sequences, which is particularly common in gliomas, but also found in lung, breast, ovarian, and other cancers. Interestingly, these deletion mutants mimic the avian erythroblastosis virus v-ErbB oncoprotein, from which the EGFR family members took their names. Numerous point mutations are also found in the kinase domain, especially in non–small cell lung cancers, which promote constitutive activation, even in the absence of ligand-induced dimerization. Again, the effects of the point mutations are also closely replicated by small insertions and deletions within the kinase domain.
Activation of the EGFR receptor results in phosphorylation of tyrosine residues in the C-terminal tail, which serve as docking sites for the recruitment of downstream effectors (see Chap. 8, Sec. 8.2). Generally, activating point mutations result in the receptor tail attaining a phosphorylation status intermediate between that of unstimulated and growth factor-stimulated wild-type receptors. An important consequence of this “partial” activation is a resulting shift in the downstream signaling output from the mutant receptors. Whereas stimulated wild-type epidermal growth factor receptors (EGFRs) activate a number of downstream pathways, the mutant receptors tend to prominently activate mitogen-activated protein (MAP) kinase and phophatidylinositol-3 (PI3) kinase signaling at the expense of the other pathways. Normally, EGF signaling is attenuated by a feedback mechanism that involves the removal of the receptors from the cell membrane by endocytosis, followed by lysosomal degradation or recycling. Some point mutations have been found to disrupt this regulatory device, resulting in hyperactivation of the EGF pathway.
EGF signaling has an important physiological role in mammary development, and deregulation of downstream pathways feature prominently in many breast tumors (Schmitt, 2009). The ERBB2/ERBB3 heterodimeric receptor complex is a potent signal transducer, particularly of the PI3 kinase pathway, because the ERBB3 receptor recruits the PI3 kinase directly rather than through intermediary adaptor proteins. The ERBB2(HER-2) gene is amplified or overexpressed in roughly one-quarter of all human breast cancers, as well as a smaller percentage of ovarian and gastric cancers. Overexpression of the ERBB2 receptor has also been correlated with elevated ERBB3 levels. Although ERBB2 somatic mutations have now been identified in some lung tumors, gene amplification is the most common mechanism for oncogenic upregulation of the receptor. Amplification of the ERBB2 gene is generally an early event in breast cancer progression and is associated with poor prognosis in breast and gastric cancers.
7.5.4 PI3 Kinase
In response to specific upstream signals, the PI3 kinases phosphorylate the 3′ hydroxyl group of phosphatidylinositols, generating membrane-embedded lipid secondary messengers that activate downstream pathways (Fig. 7–12; see also Chap. 8, Sec. 8.2.5) (Engelman et al, 2006). Although the superfamily includes a number of isoforms, the preponderance of research has focused on the Class IA PI3 kinases, which consist of p110 catalytic and p85 regulatory subunits. These preferentially convert phosphatidylinositol 4,5-bisphosphate (PIP2) to phosphatidylinositol 3,4,5- triphosphate (PIP3), which recruits Pleckstrin homology (PH) domain-containing proteins, such as the PKB/AKT serine/threonine kinase, to the plasma membrane for activation. The involvement of PI3 kinase signaling in onco-genesis was evident in the 1980s from the discovery of an interaction between the transforming polyoma viral middle T antigen and the p85 subunit of the kinase (Whitman et al, 1985; Otsu et al, 1991). In parallel, a constitutively active version of the p110 subunit was isolated from the genome of the ASV16 avian retrovirus, which was known to transform chicken fibroblasts (Chang et al, 1997).
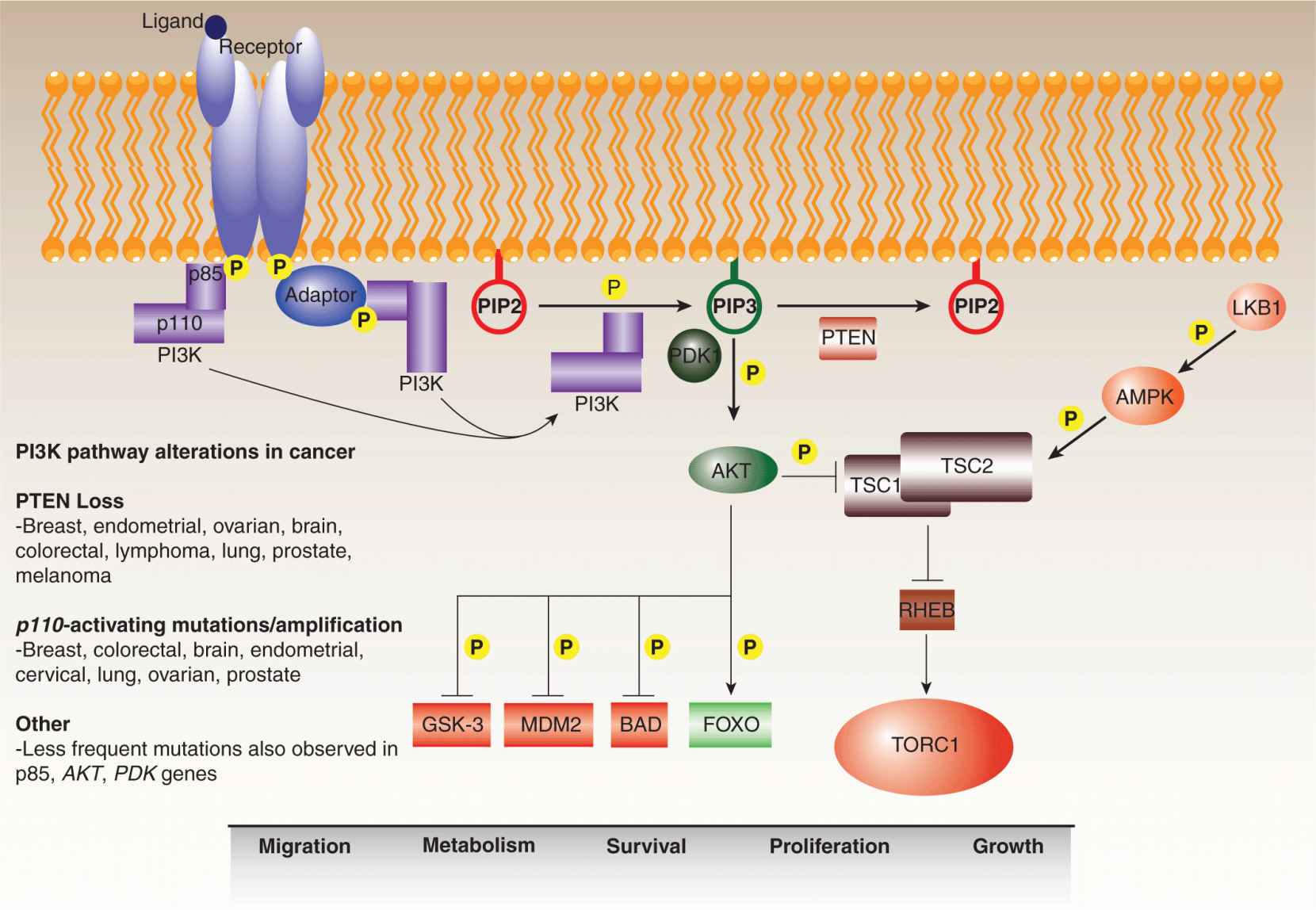
FIGURE 7–12 PI3 kinase pathway. The PI3 kinase pathway regulates a variety of cell functions, including migration, metabolism, survival, proliferation, and growth. It is frequently deregulated in human cancers at various points. In response to external stimuli, activated receptor tyrosine kinases recruit PI3 kinase via its p85 regulatory subunit, either directly or mediated by an adaptor protein such as IRS-1. Upon recruitment, activated PI3 kinase (PI3K) converts PIP2 (phosphatidylinositol 4,5 bisphosphate) to PIP3 (phosphatidylinositol 3,4,5 triphosphate). This reaction is antagonized by the lipid phosphatase, PTEN, which acts as a brake on the pathway. PH-domain proteins, such as the PDK1 and AKT kinases, are recruited to the plasma membrane by PIP3, whereupon PDK1 contributes to the activation of AKT, which then transmits the signal downstream by phosphorylating a range of targets. AKT also modulates the RHEB guanosine triphosphatase (GTPase)-TORC1 signaling axis by downregulating the TSC1–TSC2 complex, which is modulated by a number of upstream signals including activation by LKB1-AMPK in response to energy deprivation. The PI3 kinase pathway is generally overstimulated in cancers through aberrant activation of the receptors, activating mutations in the PI3 kinase catalytic subunit (p110), and through the loss of PTEN.
More recently, activating mutations have been identified in the PIK3CA gene, encoding the p110 subunit, in a number of tumors, including breast, prostate, endometrial, and colon cancers (Chalhoub and Baker, 2009). These mutations result in constitutive activation of the kinase in the absence of upstream signaling, a buildup of PIP3, and increased mobilization of downstream pathways that regulate cell processes such as growth, proliferation, survival, migration, and glucose metabolism. Remarkably, approximately 80% of all somatic PI3 kinase mutations occur at 3 hotspot residues: E542K and E545K within the helical domain, and H1047R in the kinase domain. All 3 mutations have been found to increase the in vitro activity of the kinase as well as the PI3 kinase signaling throughput in cells and tissues bearing the mutations. Amplifications of the PIK3CA gene are sometimes observed in cervical, head and neck, gastric, as well as lung cancers. Less frequent cancer-associated activating mutations have also been identified in the PIK3R gene, encoding the p85 subunit, as well as the AKT1 gene, encoding 1 of the more prominent downstream effectors of PI3 kinase signaling.
7.5.5 RAS and RAF
The importance of MAP kinase signaling in neoplastic transformation is evident from the frequency of oncogenic mutations affecting multiple proteins in the pathway, including growth factor receptors, RAS guanosine triphosphatase (GTPase), and its immediate target, RAF (Fig. 7–13) (Karreth and Tuveson, 2009). The pathway modulates a number of cellular processes, including proliferation, growth, survival, differentiation, and migration, albeit in a tissue-dependent manner (see also Chap. 8, Sec. 8.2.3). RAS genes are among the most commonly mutated genes in human cancer, with a variety of aberrations detected in at least 30% of all tumors. Among the 3 RAS genes, the preponderance of mutations is observed in the K-RAS gene, especially in pancreatic, lung, colon, endometrial, and ovarian tumors. Mutations in the H-RAS gene are more commonly observed in bladder cancers with N-RAS mutations found in neuronal, melanoma, and myeloid malignancies.
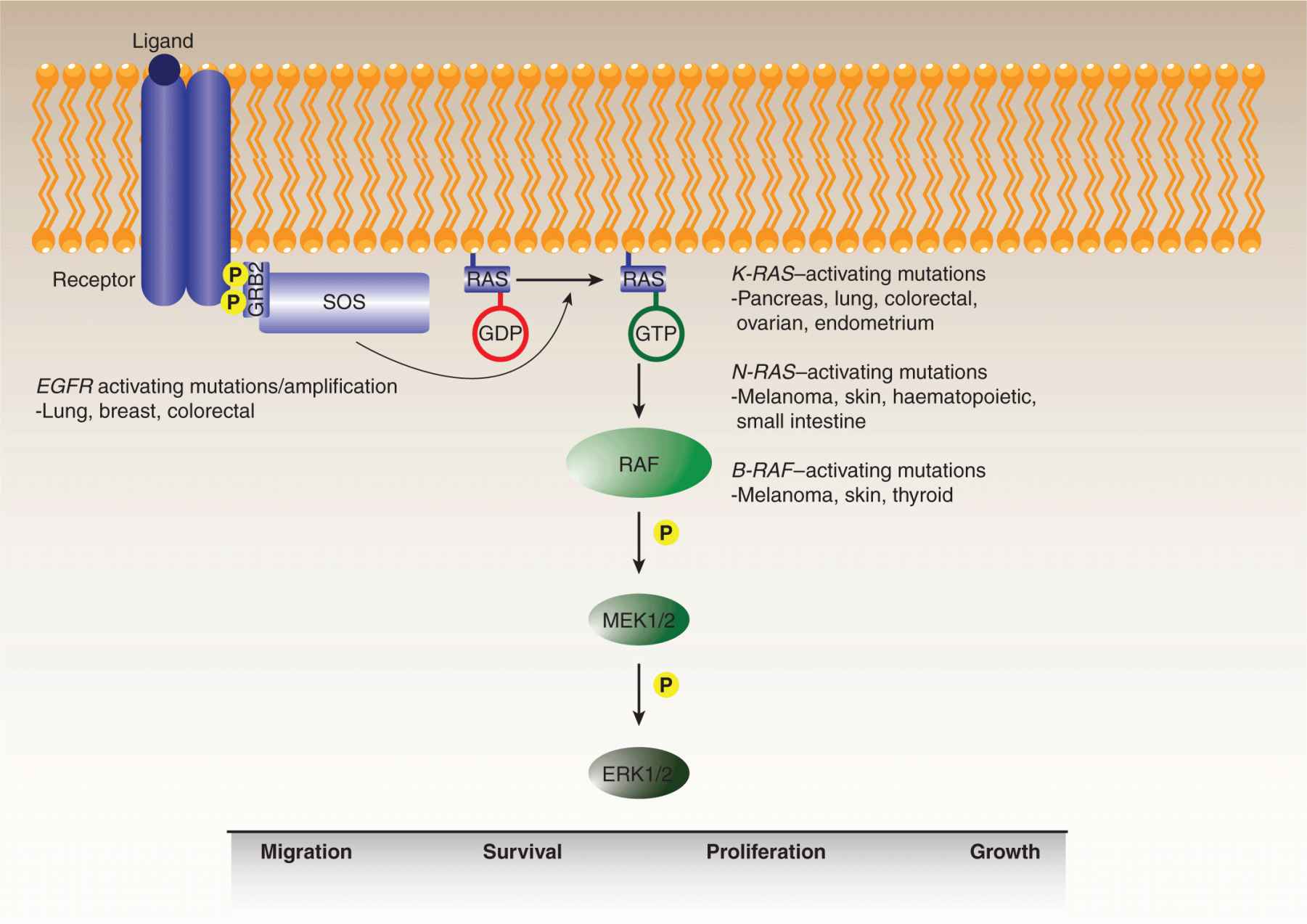
FIGURE 7–13 MAP kinase pathway. The mitogen-activated kinase (MAPK)/ERK pathway is frequently deregulated during oncogenesis. The pathway is stimulated by binding of ligands to their cognate receptors, ultimately resulting in activation of the ERK1/2 kinase. Activated receptors recruit adaptors, such as GRB2, and guanine nucleotide exchange factors (GEFs), such as SOS, which promotes the guanosine triphosphate (GTP) loading of RAS. RAF is then activated by RAS-GTP, triggering a kinase cascade resulting in the sequential activation of MEK1/2 and ERK1/2, whose downstream targets regulate a number of cell functions including migration, survival, proliferation, and growth. Overactivation of the pathway through activating mutations or amplification of receptors, as well as activating mutations in various RAS isoforms or B-RAF, is commonly observed in human cancers.
The most common mutations are gain-of-function substitutions in codons 12, 13, and 61, with many lower-frequency mutations identified throughout the protein sequence. The RAS gain-of-function mechanism is somewhat distinct in that it is the RAS GTPase enzymatic turnover that is blocked by the oncogenic mutations, effectively locking the protein into the activated guanosine triphosphate (GTP)-bound signaling state, and resulting in the upregulation of its downstream effectors.
The primary downstream effectors of RAS are the 3 RAF family serine/threonine kinases (see also Chap. 8, Sec. 8.2.3). Although a virally transduced version of C-RAF exhibits oncogenic properties, missense mutations are infrequently observed in the C-RAF gene, and these rare substitutions do not generally appear to stimulate kinase activity. Similarly, mutations in the A-RAF gene have been detected very rarely. B-RAF is the most frequently mutated RAF family member with approximately 40% of malignant melanomas and 2% of all human cancers, including thyroid, colon, and ovarian cancers, exhibiting B-RAF mutations (Beeram et al, 2005; Maurer et al, 2011). The V600E substitution in the kinase activation loop accounts for roughly 90% of all B-RAF mutations. Development of vemurafenib, an agent that specifically targets this mutation, has led to marked improvement in outcome for patients with malignant melanoma (Chapman et al, 2011). Interestingly, mutations in the RAS and B-RAF genes are almost entirely mutually exclusive, indicative of the potency of a single oncogenic hit at either gene that evidently relieves virtually all the selective pressure for additional mutations in the pathway. Indeed, the B-RAFV600E mutant protein is constitutively activated and essentially independent of upstream signaling from growth factor receptors or RAS. An analysis of the effects of the V600E substitution on the B-RAF structure suggested that the mutation results in the activation loop adopting an open conformation than is more accessible to substrates. Typically, this conformation change is accomplished by specific regulatory phosphorylation events particularly within the kinase activation loop.
7.5.6 IDH
Despite extensive genome-wide sequencing efforts to produce a detailed genetic map of the cancer landscape, surprisingly few novel oncogenes and tumor-suppressor genes have been identified. The IDH1 and IDH2 genes, encoding 2 previously unglamorous isocitrate deydrogenase metabolic enzymes, are the most prominent examples of the worthiness of these large-scale projects. The mechanism by which these proteins contribute to tumorigenesis, though far from clear, appears to define a novel oncogenic paradigm. Now recognized as onco-genes, they were initially mistaken for tumor suppressors because the observed cancer mutations destroy their normal cellular activities.
Exon sequencing carried out on human glioblastoma multiforme, as well as other cancers, revealed somatic mutations in IDH1 and IDH2 at homologous arginine positions in some of the sampled tumors (Parsons et al, 2008; Yan et al, 2009). Functional characterization of the mutant proteins indicated that the amino acid substitutions led to a loss of function, destroying their ability to convert isocitrate into α-ketoglutarate, and suggesting that these were novel tumor suppressors. However, there was no evidence of a LOH at the IDH loci in human tumors, indicating that they did not conform to Knudson’s 2-hit tumor-suppressor model. An alternative explanation for these findings arose from the observation that cells harboring the tumor-associated IDH1 mutation exhibit an accumulation of a particular metabolite, 2-hydroxyglutarate (2-HG) (Dang et al, 2009). An analysis of the effects of the mutation on the IDH1 structure suggested that although the active site of the mutant enzyme failed to interact with isocitrate, its normal substrate, it was capable of interacting with α-ketoglutarate and converting it to 2-HG. Thus, the arginine substitution effectively represents both a loss-of-function and gain-of-function mutation that destroys the normal enzymatic activity while creating a novel one.
On the basis of these results, it has been proposed that 2-HG represents an oncometabolite whose ability to promote onco-genesis needs to be fully elucidated. In addition to gliomas, IDH1 and IDH2 mutations have also been detected in AML genomes, including a novel mutation in the IDH2 sequence which also leads to 2-HG build up (Balss et al, 2008; Ward et al, 2010). Importantly, knockdown of IDH1 and IDH2 reduced the growth of cultured AML cells, consistent with their roles as oncogenes rather than tumor-suppressor genes.
7.6 LOSS OF FUNCTION EVENTS
7.6.1 p53
The most extensively studied tumor-suppressor gene is the p53 transcription factor, which was discovered in 1979 as an interacting partner of the SV40 viral T-antigen (Brosh and Rotter, 2009). In response to cellular stresses, a variety of mechanisms converge on p53 leading to its stabilization and accumulation (Junttila and Evan, 2009). Contingent upon the extent of the stress signals, p53 then activates the expression of genes involved in DNA repair and growth arrest on the one hand, or apoptotic cell death on the other (Fig. 7–14). In the absence of p53, cells are impaired in their ability to appropriately respond to oncogenic stresses, leading to tumorigenic proliferation, growth, and genetic instability. Remarkably, despite the vast amount of p53 research, new functions for this multitasking protein are still being discovered. Somatic mutations in the TP53 gene, encoding the p53 protein, are found at varying frequencies in many human cancers, ranging from roughly 10% in some leukemias to more than 70% in advanced lung carcinomas. The majority of mutations in the TP53 gene are missense substitutions, with the remainder consisting of frameshift, nonsense, and silent mutations, along with rare deletions and insertions. In addition to somatic changes, germline p53 mutations lead to Li-Fraumeni syndrome, a highly penetrant autosomal dominant disorder that causes tumor growth in a wide spectrum of tissues. For both somatic and germline mutations, oncogenic progression almost always involves LOH at the p53 locus.
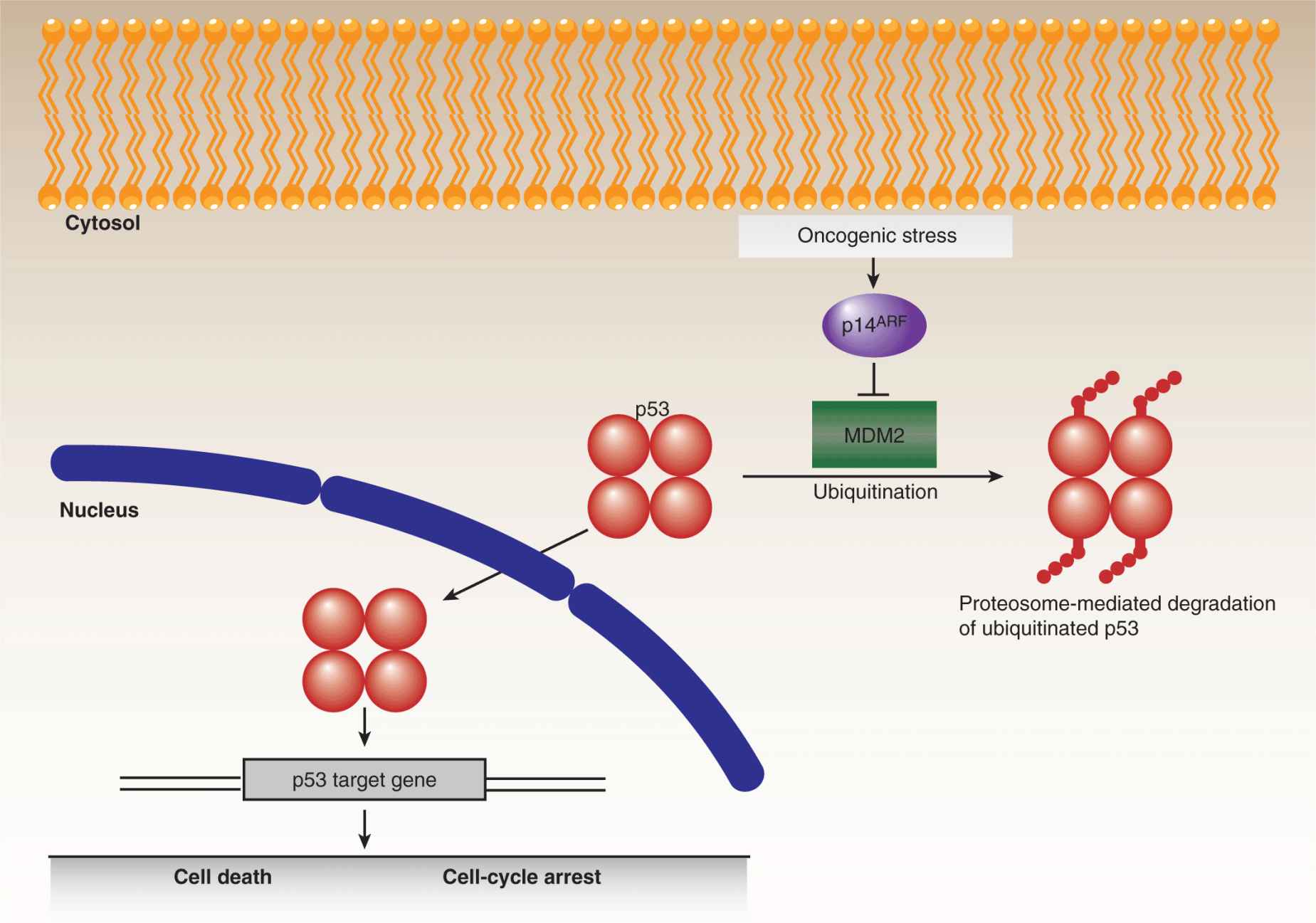
FIGURE 7–14 p53 and cell fate determination. The p53 tumor suppressor represents a critical regulatory node for cell fate determination and is the most frequently mutated gene in human cancers. The levels of p53 are normally constrained, primarily by the action of MDM2, which promotes the ubiquitination and proteasomal degradation of the protein. In response to cellular stresses, p53 is stabilized and translocates to the nucleus and regulates gene expression. Depending on the level of stress, p53 can promote different cellular outcomes, notably cell-cycle arrest, to permit the alleviation of the particular stress or, in irretrievable cases, cell death. Because oncogenic stresses can inhibit MDM2, through the activity of p14ARF, tumorigenic progression requires either the circumvention or lo ss of p53 activity.
Although thousands of p53 mutations have been cataloged, 6 “hotspot” residues, located in the central DNA binding domain, are mutated in a disproportionate percentage of p53-associated cancers. Two of these mutations, R248Q and R273H, affect p53 activity by interfering with its capacity to bind DNA, while the other mutations, R249S, G245S, R175H, and R282W, cause varying degrees of structural disruption. The traditional models of p53 function, in relation to oncogenesis, involve a combination of loss-of-function and dominant-negative effects, the latter mediated by dimerization of mutant p53 with wild-type p53 (thereby inhibiting normal p53 function), prior to LOH. The inability of mutant p53 to bind DNA naturally results in reduced expression of p53 targets, including the gene that encodes the MDM2 ubiquitin ligase (see Chap. 8, Fig. 8–3 and Chap. 9, Fig. 9–4), which strongly influences the turnover of p53 itself. As a consequence, mutant p53 levels are frequently, although not always, elevated in tumors, enhancing the dominant-negative effect. More recently, it has been proposed that some p53 mutations confer gain-of-function oncogenic effects, at least partly involving inactivation of the other p53 family members, p63 and p73, as well as some unrelated transcription factors. Researchers are now designing drugs (eg, PRIMA1) that selectively target cancers with mutated p53 by promoting conversion of the mutant conformation to the wild-type form, or by driving downstream effector proteins (eg, MDM2) to reengage apoptotic pathways, thereby sensitizing tumors to chemotherapy and radiotherapy (Brown et al, 2009).
7.6.2 PTEN
Initially identified during the 1990s as the major tumor suppressor at chromosome 10q23, PTEN was found to directly counter PI3 kinase signaling by acting as a lipid phosphatase for PIP3, the product of PI3 kinase activity (see Fig. 7–12) (Chalhoub and Baker, 2009; Hollander et al, 2011). Down-regulation of PTEN correlates with increased PIP3 levels and hyperactivation of downstream PKB/AKT-mediated signaling. Although reported initially as an exclusively cytoplasmic enzyme, there is also a nuclear PTEN cohort in at least some cells. The nuclear functions of PTEN have not yet been fully documented, though, intriguingly, PTEN loss appears to correlate with genomic instability. The protein structure consists of a short N-terminal lipid-binding domain followed by the phosphatase domain, a membrane-binding C2 domain, and a C-terminal tail that influences stability and subcellular localization.
Germline PTEN mutations are causal in the related Cowden, Bannayan-Riley-Ruvalcaba, Proteus, and Proteus-like hamartoma syndromes, collectively known as PTHS (PTEN hamartoma tumor syndromes). These clinically distinct conditions are marked by small benign growths and, in Cowden syndrome, carry an enhanced risk for developing breast, thyroid, and endometrial cancers. Importantly, progression from benign to malignant tumor growth, in Cowden syndrome, coincides with LOH at the PTEN locus.
Sporadic PTEN loss-of-function, effected at the genomic, epigenomic, transcriptional, or protein levels, is found in a wide array of cancers, especially glioblastomas, melanomas, prostate, breast, and endometrial tumors. For example, genomic deletions at the PTEN locus have been observed in roughly 70% of glioblastomas, with biallelic inactivation occurring in roughly 25% to 40% of these malignancies. PTEN deletions are also observed in premalignant endometrial lesions, suggestive of a role in the initiation of endometrial carcinomas. In addition to missense substitutions, nonsense and frameshift mutations have also been observed, yielding truncated mutants that retain the phosphatase and C2 domains, but not the more C-terminal regions. These mutants lack the capacity to suppress the growth of PTEN-deficient cells, indicating a deficiency in regulation or stability. Silencing of the PTEN promoter by methylation has been found in some thyroid and lung cancers, as well as in melanomas and glioblastomas.
Beyond genomic and epigenomic disruptions, PTEN is also downregulated by oncogenic mechanisms operating at other regulatory levels. The miR-21 microRNA, which promotes tumorigenesis by targeting the PTEN transcript, is overex-pressed in a wide array of cancers, including hematological malignancies such as AML and CLL, along with solid tumors such as glioblastomas, breast, colon, and lung cancers (Leslie and Foti, 2011). The PICT-1 protein, which stabilizes PTEN by interacting with its C-terminal tail, is lost in some neuroblastomas, and PICT-1 deficiency correlates with PTEN downregulation at the protein level. The critical role played by PTEN in suppressing tumorigenesis in many tissues is underscored by the variety of means through which its function is controlled.
7.6.3 BRCA1 and BRCA2
In 1994, linkage analysis studies correlated the BRCA1 and BRCA2 genes with hereditary breast cancer and hundreds of germline and somatic mutations have since been identified in both genes (Narod and Foulkes, 2004). BRCA1 mutations associate with high-grade infiltrating ductal carcinomas, frequently exhibiting a basal, triple-negative phenotype (ie, without expression of hormone receptors or HER-2/ERBB2; see Chap. 20, Sec. 20.3), whereas BRCA2 mutations do not appear to correlate with a particular tumor histopathology. In addition to breast cancer, BRCA1 and BRCA2 carriers are now also known to bear a heightened risk for developing ovarian tumors, and male carriers of BRCA2 have an increased risk of prostate cancer.
The BRCA1/2 tumor suppressors play central roles in the maintenance of genomic integrity (Huen et al, 2010). The BRCA1 structure consists of an N-terminal RING domain, a central coiled-coil domain, and a C-terminal BRCT (BRCA1 C-terminus) domain, all of which mediate interactions critical to the functions of the protein in the realms of DNA repair, cell-cycle checkpoint control, chromatin remodeling, and ubiquitination. The RING domain, which interacts with the BARD1 protein to create an E3 ubiquitin ligase holoenzyme, is a common target for oncogenic mutations. Frequent mutations are also found in the coiled-coil and BRCT domains, which disrupt BRCA1 function by interfering with protein–protein interactions. BRCA1 is involved in the resection step of DNA double-stranded break (DSB) repair by homologous recombination, via its interaction with the MRN endonuclease complex (see Chap. 5, Sec. 5.3.4). Moreover, BRCA1 also appears to modulate DNA damage-triggered cell-cycle checkpoints by regulating activation of the Chk1 kinase (see Chap. 9, Sec. 9.3.1).
BRCA2 is a large protein containing several regions known to mediate interactions, notably 8 central BRCT repeats that bind RAD51 and C-terminal nucleic acid recognition sequences (known as OB folds) that interact with single-stranded DNA (ssDNA). Recently, 3 groups reported the successful purification of the BRCA2 protein, enabling greater mechanistic insights (Jensen et al, 2010; Liu et al, 2010; Thorslund et al, 2010). The primary function of the protein is in facilitating DNA repair by homologous recombination through the recruitment of RAD51 to the resected ssDNA stretches generated by the MRN complex at sites of DNA damage (see Chap. 5, Sec 5.3.4). BRCA1 is also involved in this phase of DNA repair via its interaction with BRCA2. Tumors which lack functional BRCA1 or BRCA2, and therefore are deficient in homologous recombination, can be selectively targeted by inhibition of poly(adenosine diphosphate-ribose) polymerase (PARP) which provides an alternative pathway for DNA repair; this is the prototype example of the use of “synthetic lethality” in the treatment of cancer (see Chap. 5, Sec. 5.8 and Chap. 17, Sec. 17.3.2).
7.6.4 Retinoblastoma Protein
The Rb protein is an important transcriptional regulator, which is inactivated in a substantial portion of human cancers, beyond the historic association alluded to in its name. In part, Rb protein acts as a cofactor interacting with and regulating the activity of transcription factors, notably those of the E2F family. In addition, Rb protein serves as an adaptor for the recruitment of epigenetic regulators, thereby impacting the chromatin status both at specific loci and at a global level. Originally, the tumor suppressor function of Rb protein was attributed largely to its role in constraining cell-cycle progression, at the G1 to S phase transition, by influencing the expression of genes under the control of E2F transcription factors (see Chap. 9, Sec 9.2.3). In its hypophosphorylated form, Rb binds E2F proteins and prevents them from activating target genes required for cell-cycle progression. In response to proliferative signals, CYCLIN/CDK complexes phosphorylate Rb protein resulting in its dissociation, thereby liberating the E2F proteins to activate the appropriate gene expression program. It is now appreciated that Rb protein also affects the expression of transcriptional targets that impact a wide array of cellular processes encompassing differentiation, cell death, genomic stability, senescence, and angiogenesis. Deregulation of any of these through Rb protein inactivation would clearly have the potential to promote various stages of tumorigenesis, although the precise Rb protein contribution likely varies in different tumors.
7.7 THERAPEUTIC STRATEGIES GUIDED BY CANCER GENETICS
The treatment of cancers with radiation and conventional chemotherapy largely entails broad-spectrum killing of both malignant and untransformed cells resulting in varying levels of toxicity. The genetic heterogeneity of cancer, although complex, presents oncologists with an opportunity for more targeted approaches. The prospect of personalized medicine is now a tangible objective, encompassing the capacity to profile the totality of aberrations in individual tumors, and then tailor treatments to maximize the targeted killing of cancer cells while reducing attendant side effects (see Table 7–1). The advent of high-throughput screening technologies has enabled the timely identification of promising targeted therapeutics from libraries of small-molecule compounds. In parallel, humanized monoclonal antibodies have been developed to interfere with oncogenic pathways at the cell surface, for example, by blocking ligand binding or tagging cancer cells for immunological clearing.
Diagnostic tests for profiling genetic changes in human tumors have become commercially available and many more are anticipated. One class of diagnostic assays scans tumor DNA for hundreds of known cancer-associated mutations. Currently, this is accomplished by high-throughput chip-based mass spectrometry platforms such as the Sequenom MassArray system, which has been used to screen panels of genes including the OncoCarta and OncoMap sets (MacConaill et al, 2009; see Chap. 2 for description of techniques). Single-nucleotide mutations, insertions, deletions, and copy number variations, as well as epigenetic changes, can all be detected with this technique. As costs are reduced by the development of next-generation DNA sequencing methods, more comprehensive probing of tumor DNA alterations will soon be available (Ansorge, 2009).
A second class of diagnostic tests is based on the profiling of tumor gene expression patterns in order to predict clinical scenarios, such as metastatic propensity, disease recurrence, and therapeutic sensitivity (Nevins and Potti, 2007). One such assay is the MammaPrint microarray test that analyzes a 70-gene signature that is predictive of metastatic spread in early stage breast tumors (van ‘t Veer et al, 2002). Another example, is the Oncotype DX test in which the expression levels of 21 genes are assessed by reverse-transcription polymerase chain reaction (RT-PCR) and used to predict the probability of 10-year recurrence and responsiveness to adjuvant chemotherapy in patients with early stage breast cancer (Sparano and Paik, 2008; see Chap. 20, Sec. 20.3.2). A similar test, based on a 12-gene panel, for predicting 10-year recurrence in patients with stage II colon cancer is also now available. Given the heterogeneity of genetic alterations among cancers within individual tissues, more ambitious gene expression signatures are being developed in order to distinguish tumor subtypes, thereby facilitating the design of more precise therapeutic regimens.
The discovery of the oncogenic BCR-ABL fusion protein (Sec. 7.5.1) triggered a search for compounds that could specifically inhibit the kinase. The resulting treatment of CML with the specific BCR-ABL inhibitor, imatinib, remains the benchmark for targeted cancer therapeutics. Although imatinib has proven to be highly effective in preventing the progression of CML from the chronic to blast phases, secondary mutations in the BCR-ABL kinase domain can lead to the development of therapeutic resistance. In such cases, successful management of the disease has involved countering the secondary resistance with other tyrosine kinase inhibitors, such as dasatinib. Because CML is associated with a specific genetic lesion, it is debatable whether it can truly be regarded as a paradigm for countering the progression of tumors dependent on perhaps 15 driver mutations. Such tumors are unlikely to have a single “Achilles’ heel” that can be specifically targeted, and cocktails of targeted therapies will likely be required. Moreover, recent sequencing efforts have confirmed that spatially distinct regions of individual tumors are often occupied by subclonal populations of tumor cells whose genomes have acquired divergent sets of driver mutations, in addition to the common mutations present in the original clonal population. This intratumor heterogeneity also would be expected to limit any therapeutic strategy that depended on a single agent.
Several targeted therapeutics have been designed to counter hyperactivation of EGF pathways. Non–small cell lung cancers (NSCLCs), often driven by EGFR hyperactivation, have been treated with the specific EGFR inhibitors, erlotinib and gefitinib (Pines et al, 2010). Both are effective against only a subset of targeted malignancies, and this initial potency is commonly followed by the development of therapeutic resistance. Gaining an understanding of the molecular factors underlying both the initial insensitivity and acquired resistance is indispensable for guiding modifications to the therapeutic regimens.
Small-molecule EGFR inhibitors have generally proved disappointing in treating refractory colorectal cancers, although EGFR pathway activation is often observed in these tumors. Cetuximab, a humanized monoclonal antibody that binds to the EGFR ligand-binding domain promoting cytosolic internalization of the receptor, has demonstrated some clinical efficacy. Improved therapeutic response has been observed, mostly in combinatorial treatment regimens, in trial settings evaluating cetuximab as both first-line therapy, as well as in the treatment of refractory colorectal cancers. Moreover, evaluation of trial data to identify predictive biomarkers determined that tumors bearing K-ras mutations were not responsive to cetuximab, presumably because of downstream activation of the EGFR pathway.
ERBB2 (HER-2) status is an important predictive marker, in either early stage or metastatic breast cancers, for tumor sensitivity to treatment with trastuzumab, a humanized monoclonal antibody that targets the ERBB2 ectodomain (Finn and Slamon, 2003). Mammary carcinomas are screened for ERBB2 status by a combination of immunohistochemistry (IHC), to gauge receptor levels at the cell surface, and fluorescence in situ hybridization (FISH) to establish gene copy numbers. A finding of greater than 6 ERBB2 gene copies is suggestive of a tumor that may be receptive to treatment with trastuzumab. However, a substantial portion of tumors with high ERBB2 status prove to be refractory to the therapy, presumably because of additional oncogenic events, such as loss of the PTEN tumor suppressor, which obviates the requirement for upstream signals from the ERBB2 receptor for downstream pathway activation.
In light of the frequent upregulation of MAP kinase and PI3 kinase signaling in a wide array of cancers, the various actors in the pathway constitute attractive therapeutic targets. However, the clinical performance of drugs targeting these pathways has been minimal, although new compounds are being evaluated in trials. RAS hyperactivation is somewhat paradoxical in that oncogenic mutations lock the protein in an activated state by blocking enzymatic turnover. This poses a problem for drug designers who typically generate compounds designed to block the enzymatic catalysis, a conceptually simpler task for which established inhibitory strategies are known.
Deactivation of RAS requires stimulation of enzymatic turnover and, despite great effort, there is no drug capable of effectively downregulating oncogenic RAS. In addition to GTP binding, RAS activation involves membrane translocation brought about by the multistep covalent attachment of specific lipid groups near the C-terminus of the protein. Drugs, such as farnesyltransferase inhibitors (FTIs), designed to disrupt this lipidation process, have either proven ineffectual or overly toxic (Beeram et al, 2005). The prospect of interfering with the MAP kinase pathway downstream of RAS, using RAF or MEK inhibitors, is being pursued. RAF inhibitors have proven to be potent against cancers driven by the oncogenic B-RAFV600E mutant, such as malignant melanoma, but, counterintuitively, have demonstrated tumorigenic properties when treating experimental cancers harboring oncogenic RAS or the wild-type RAS/RAF combination (Heidorn et al, 2010; Chapman et al, 2011). This finding is indicative not only of the complexity of MAP kinase signaling, but also of the careful consideration which must be accorded to the full genetic context of a cancer prior to therapeutic administration. MEK inhibitors are also being assessed, although there is evidence that some RAS mutant tumors do not require MEK activation for oncogenic signaling.
Although the PI3 kinase inhibitors, Wortmannin and LY294002, have been used in research for the better part of 2 decades, a combination of poor pharmacological properties and a lack of specificity have hampered clinical applications. Despite these challenges, the prevalence of the pathway in neoplastic transformation has encouraged continued investment into the creation of drugs targeting specific PI3 kinase isoforms, and a number of compounds are at different stages of clinical development.
Many promising targets, such as RAS and MYC, may prove to be “undruggable,” necessitating alternative attack strategies. Synthetic lethal studies can be exploited to uncover secondary targets, more amenable to therapeutic intervention, whose inhibition can block cancer progression driven by specific oncogenic pathways. As noted, there is interest in the use of PARP inhibitors in patients whose tumors have lost expression or function of the BRCA1 and BRCA2 DNA repair genes, and become dependent on PARP to repair DNA. Still other targets will prove promising in one genomic context, but when inhibited in the presence of other mutations may have limited effect on tumorigenesis. As noted, the targeting of upstream receptors is ineffectual when additional mutations in the K-RAS, B-RAF, PIK3CA, or PTEN genes activate the downstream pathways irrespective of receptor signaling. Granting these qualifications, the first step in dealing with a given cancer will be to define the nature of the oncogenic transformation through timely and cost-effective profiling of the tumor.
Related posts:
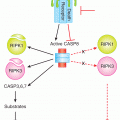
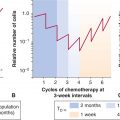
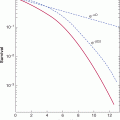
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


