Figure 2-1 Receptor tyrosine kinase signaling in cancer Scheme for growth factor signaling through receptor tyrosine kinases.
Oncogenes
Oncogenes encode proteins that possess the ability to cause cellular transformation. These genes act in a dominant fashion, through either overexpression or activating mutations. There are several criteria that define cellular transformation. These include morphological changes, loss of contact inhibition, anchorage-independent growth, and the ability to form tumors when transplanted into nude mice. For example, under normal physiological situations, a growth factor binding to a receptor produces a very transient activation of a certain signaling cascade allowing tightly regulated responses such as proliferation to occur. When downstream components of these cascades are mutated in a way that causes them to be constitutively active, the signal is no longer transient and regulated but is aberrantly turned on in a continuous fashion. In addition to activating mutations, these genes can be activated by overexpression at levels much higher than in normal cells. Proto-oncogenes are commonly involved in cellular signaling, and specific examples are discussed later in the context of their roles in signal transduction.
Initially, it was believed that cellular transformation was caused solely by unregulated cell proliferation induced by activation of oncogenes. It is now known that although deregulated proliferation is most likely a necessary component for transformation, it is probably not sufficient, and other changes, such as modulation of cell survival functions, are critical as well. In fact, as discussed later, certain oncogenes function to modulate cell survival.
In the early 1980s, approaches aimed at identifying the functions of retroviral oncogenes converged with efforts to investigate normal mitogenic signaling by growth factors. Analysis of the predicted sequences of a number of retroviral oncogene products uncovered several with similarities to the prototype v-src product, whose enzymatic function as a protein kinase had been identified. Unlike many protein kinases, which phosphorylated serine and/or threonine residues, the v-src product was a protein kinase capable of specifically phosphorylating tyrosine residues. 2 Later efforts to identify oncogenes led to the discovery of the small GTPase Ras, which was unmasked as a transforming gene by transfection of tumor cell genomic DNA. 3
Independent efforts to purify and sequence growth factors led to the discovery that the sequence of the platelet-derived growth factor (PDGF) B chain matched the predicted product of the transforming gene of simian sarcoma virus, designated v-sis. 4,5 The v-erbB gene of avian erythroblastosis virus, which predicted a v-src–related protein tyrosine kinase, was then found to represent a truncated form of the epidermal growth factor receptor (EGFR). 6 Independent evidence demonstrated that EGF triggering of its receptor resulted in tyrosine autophosphorylation. 7 Thus, a direct link between growth factors, receptors with tyrosine kinase activity, and oncogenes was firmly established. The proliferation, differentiation, functional activity, and survival of cells can be affected by a wide array of other cytokines that signal through transmembrane receptors that lack protein tyrosine kinase activity. Because these signaling systems have also been implicated in malignant transformation, they are described in this chapter as well.
Signal Transduction by Protein Tyrosine Kinase Receptors
Membrane-spanning RTKs contain several discrete domains, including their extracellular ligand binding, transmembrane, juxtamembrane, protein tyrosine kinase, and carboxy-terminal tail domains. 8,9 Interaction of a growth factor with its receptor at the cell surface leads to a tight association, so that growth factors are capable of mediating their activities at very low concentrations. In the general model of RTK activation, ligand binding induces receptor dimer or oligomer formation associated with activation of the tyrosine kinase domain. However, the dimerization mechanism can be very different among the 20 families of RTKs, and the contribution of the ligands varies. For example, nerve growth factor (NGF) dimers directly mediate the coupling of two TrkA receptors, whereas EGF molecules do not contribute to the EGFR dimerization interface, but instead induce a conformational switch, which unmasks a dimerization domain in the EGFR extracellular region. 9 Also, it should be noted that certain RTKs, such as the insulin and insulin-like growth factor receptors, form inactive dimers that are triggered in the presence of their ligands through conformational changes. 9
Among the different RTKs, the tyrosine kinase is the most conserved domain, and its integrity is absolutely required for receptor signaling. For example, mutation of a single lysine in the ATP binding site, which blocks the ability of the receptor to phosphorylate tyrosine residues, completely inactivates receptor biological function. 10 In the presence of their ligands, the conformational changes in the activated receptors overcome cis-autoinhibitory interactions in the kinase, the juxtamembrane, and the carboxy-terminal domains, which allow trans-phosphorylation on tyrosine residues that serve as docking sites for different adaptor and second messenger proteins. 9
The juxtamembrane sequence that separates the transmembrane and cytoplasmic domains is not well conserved among different families of receptors. However, juxtamembrane sequences are highly similar among members of the same family, and studies indicate that this stretch plays a role in the modulation of receptor function. For example, addition of PDGF to many types of cells causes a rapid decrease in high-affinity binding of EGF to its receptor, which is mediated by the phosphorylation of the EGFR juxtamembrane domain induced by PDGF-triggered protein kinase C (PKC), a process termed receptor transmodulation. 11 The carboxy-terminal domain of the receptor is thought to play an important role in the regulation of kinase activity. This region typically contains several tyrosine residues, which are autophosphorylated by the activated kinase. In fact, the receptor itself is often the major tyrosine phosphorylated species observed following ligand stimulation. Tyrosine phosphorylation of the carboxy-terminal domain can modulate kinase catalytic activity and/or the ability of the kinase to interact with substrates. Thus, mutations that alter individual tyrosine sites or deletions of this domain have the effect of attenuating kinase function. 9,11
RTKs and Cancer
The constitutive expression of a growth factor and its specific receptor by the same cell may be sufficient to establish a so-called autocrine loop that contributes to tumor progression. Autocrine transforming interactions have been identified in a number of human malignancies. At least one PDGF chain and one of its receptors have been detected in a high fraction of sarcomas and in glial-derived neoplasms. 12–14 Growth factors also contribute to tumor progression by a paracrine mode. For example, continuous stimulation by growth factors in paracrine as well as autocrine modes during chronic tissue damage and repair associated with cirrhosis and inflammatory bowel disease may predispose to tumors. 15 Some tumor cells produce angiogenic growth factors such as the vascular endothelial growth factors (VEGFs). Such growth factors cause paracrine stimulation of endothelial cells, inducing neoangiogenesis and lymphangiogenesis, which contribute to tumor progression. 16
RTKs are frequently targets of oncogenic alterations, which create a constitutively activated receptor, independent of the presence of ligand. This was initially demonstrated with retroviral oncogenes, v-erbB and v-fms, encoding activated forms of the EGF and CSF-1 receptor, respectively. 6,17 Alterations affecting a large number of RTKs have been implicated in human malignancies. One mechanism involves the amplification or overexpression of a normal receptor. Examples include EGFR, ERBB-2, and MET (see reviews 18,19 ). In some human tumors, deletions within the external domain of the EGFR receptor or mutations in its tyrosine kinase domain are associated with its constitutive activation. 19 The RET gene is activated by rearrangement, as a somatic event, in about one third of papillary thyroid carcinomas. Germline mutations affecting the cysteine residues in the extracellular region, resulting in constitutive dimerization of these receptors caused by the formation of intermolecular cysteine bridges, are responsible for multiple endocrine neoplasia (MEN) 2A and for the familial medullary thyroid carcinoma syndrome. In contrast, point mutations in RET kinase domain, such as those involving codons V804, Y806, A883, and M918, are responsible for MEN 2B. 20 These mutations have been shown to upregulate RET catalytic function, resulting in its genetic transmission as an oncogene. MET is overexpressed and/or mutationally activated in a variety of human tumors, including hepatocellular, gastric, and colorectal carcinomas, and activating mutations of MET have been associated with the metastatic progression of head and neck cancers. 18 A direct role of MET in hereditary papillary renal carcinoma has also been established. 21 This hereditary disease is characterized by multiple, bilateral renal papillary tumors, in which mutations activate constitutive kinase activity and transforming properties. Somatic mutations in MET have also been detected in some sporadic renal papillary tumors. 21 Several other receptors, including PDGF-β, TrkA, TrkC, and anaplastic lymphoma kinase (ALK) have been shown to be oncogenically activated in human malignancies by gene rearrangements that lead to fusion products containing the activated TK domain. 22–25
Signaling Pathways of Tyrosine Kinase Receptors
Knowledge of the cascade of biochemical events triggered by ligand stimulation of tyrosine kinase receptors has increased rapidly in recent years and provides further evidence of the importance of these signaling pathways in cancer. The PDGF system has served as the prototype for identification of the components of these systems. Certain molecules become physically associated and/or phosphorylated by the activated PDGF receptor kinase. Those identified to date include phospholipase C (PLC)-γ, 26 phosphatidylinositol-3’-kinase (PI-3K) regulatory subunit (p85), 27 Nck, 28 the phosphatase SHP-2, 29 Grb2, 30 Crk, 31 ras p21 guanosine triphosphatase (GTPase)-activating protein (GAP), 32,33 and src and src-like tyrosine kinases. 34 PLC-γ is one of several PLC isoforms and is involved in the generation of two important second messengers, inositol triphosphate (IP3) and diacylglycerol (DAG), which cause release of stored intracellular calcium and activation of PKC, respectively. 35,36 PKC participates in cell proliferation, survival, and migration. Although different isoforms can play distinct and sometimes opposite roles in such processes, evidence for the involvement of PKC in tumorigenesis and cancer progression is substantial. 36
PI-3K phosphorylates the inositol ring in PI in the 3’ position and becomes physically associated with a number of activated tyrosine kinases. 37,38 This protein contains an 85-kDA regulatory subunit, which is tyrosine phosphorylated, and a 110-kDa catalytic subunit. PI3-K appears to play a major role in cell survival signaling, as discussed later (see Figure 2-1).
RAS
Ras small GTP-binding proteins are a major point of convergence in receptor tyrosine kinase signaling and are an important component of the cellular machinery necessary to transduce extracellular signals (see review 39 ). These membrane-bound intracellular signaling molecules mediate a wide variety of cellular functions, including proliferation, differentiation, and survival. The Ras family of GTPases contains 39 proteins encoded by 36 genes in the human genome; it includes H-, N-, and K-Ras, R-Ras, Rap1 (A and B), TC21, and Rheb1/2. 39 Ras proteins are synthesized in the cytosol and become associated with the inner leaflet of the plasma membrane via posttranslational modifications, including a form of fatty acid lipidation, isoprenylation, on Cys-186. The carboxy-terminal CAAX box (Cys, two aliphatic amino acids, followed by any residue) is an essential motif required for Ras function, as it targets the unprocessed protein for this essential modification.
Ras proteins act as molecular switches alternating from an inactive guanosine diphosphate (GDP)-bound state to an active GTP-bound state. The paradigm for Ras activation involves the recruitment of a guanine nucleotide exchange factor (GNEF) to the membrane in response to growth factor binding and subsequent activation of a receptor tyrosine kinase. 39 GNEFs promote the release of GDP from the Ras catalytic pocket, and the relative abundance of intracellular GTP as compared to GDP ensures preferential binding of GTP (Figure 2-2 ). The best example of a Ras GNEF is SOS (son of sevenless), which is brought to the membrane by its stable association with the adaptor protein Grb2 (see Figure 2-1 , 40 ). Grb2 contains an src-homology 2 domain (SH2), which binds to a specific motif containing phosphorylated tyrosine residues on several RTKs, including the PDGFR and the EGFR. Grb2 also has two SH3 domains that mediate its binding to SOS via a carboxy-terminal proline-rich region. Alternatively, another adaptor protein, Shc, can bind to the cytoplasmic tail of the receptor through its SH2 domain, resulting in its phosphorylation on tyrosine and subsequently binding Grb2. 41 The exact sequence of binding of adaptors depends on the receptor and cell type. Once SOS is translocated to the membrane, it can promote the release of GDP from Ras, allowing GTP, which is present in excess in the intracellular environment, to bind and ultimately lead to Ras activation. Although Ras is a GTPase, its intrinsic GTPase activity is actually quite inefficient and requires additional proteins known as GTPase activating proteins (GAPs) to promote GTP hydrolysis (see Figure 2-2). GAPs can accelerate GTP hydrolysis by several orders of magnitude and are, thus, negative regulators of Ras functions. 39,42 The mechanism by which GAP accelerates the GTPase reaction is complex and not completely understood. Currently, several GAPs for Ras have been identified, including p120 GAP and NF1-GAP/neurofibromin, as well as GAPs with preferential activity on related proteins such as Rap. 42 Of particular interest is NF1, as it is found to be frequently inactivated by mutation in patients with the familial tumor syndrome neurofibromatosis type 1.
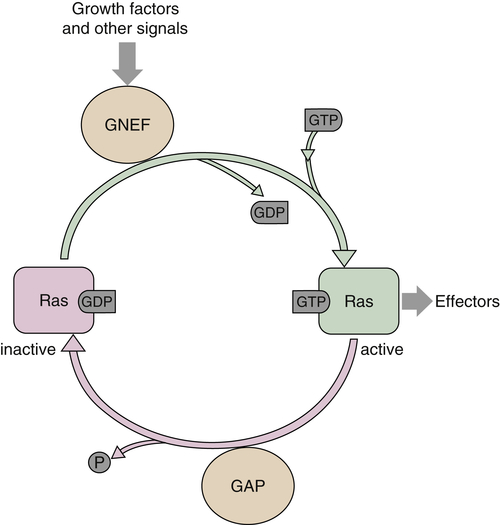
Figure 2-2 Activation of Ras GTPase
Ras Functions
Ras appears to have a multitude of functions that differ depending on factors such as cell type and extracellular environment. It is paradoxical that a single gene can cause cell cycle entry and DNA synthesis in one type of cell, such as fibroblasts, and terminal differentiation in others, such as PC12. 43,44 In other cell types such as myoblasts, activated Ras seems to oppose cell cycle withdrawal and differentiation into myotubes and downregulates expression of muscle-specific mRNA transcripts. 45 In addition, Ras has been demonstrated to promote cell survival in some cell types such as those of hematopoietic lineages on cytokine withdrawal, and PC12 cells and primary sympathetic neurons on removal of NGF or other trophic factors. 46,47 Although Ras mediates such important cellular processes as proliferation, survival, and differentiation, the exact contribution of H-, N-, and K- isoforms is not clear: targeted knockouts to H- and N‒Ras genes resulted in mice that did not exhibit an abnormal phenotype, whereas a K‒Ras knockout is an embryonic lethal and exhibits liver and hematopoietic defects. 48,49 Of note, H‒Ras knockin into the K‒Ras locus does not perturb mouse embryonic development, implying that the phenotype of K‒Ras −/− mice is probably due to the different expression pattern of the other Ras isoforms in the embryo, rather than their incapacity to compensate for K‒Ras function. 39
Ras and Cancer
The initial evidence for Ras involvement in cancer came from the discovery of transforming retroviruses, Harvey and Kirsten sarcoma viruses, which contained H- and K‒ras cellular derived oncogenes. The first human oncogenes were identified by transfecting genomic DNA from human tumor cell lines into NIH3T3 mouse fibroblasts and isolating the DNA fragments from the transformed foci. These were shown to be the human homologues of the viral ras genes. 43 Subsequent studies have shown that Ras is oncogenically activated by mutations in about 30% of all human tumors, and in some cancers, such as pancreatic carcinoma, the frequency is as high as 60%. 50
Mutations in human tumors have been found at residues 12, 13, and 61, with positions 12 and 61 being the most common. 50 These mutations decrease the intrinsic rate of GTP hydrolysis by Ras, as well as make the molecule significantly less sensitive to GAP-stimulated GTP hydrolysis. Thus, the outcome is a molecule that is predominantly GTP bound, constitutively active, and able to activate downstream pathways in the absence of growth-factor stimulation. Oncogenic Ras is capable of transforming immortalized rodent fibroblasts or epithelial cells. 43 Ras transformed cells appear refractile and spindle shaped, have disorganized actin filaments, and have a decreased affinity for the substratum. They can proliferate in the absence of adhesion (anchorage independence) or in the presence of low serum concentration. Such cells exhibit a loss of contact inhibition and grow to high saturation density. Of note, however, Ras alone is unable to transform primary mouse or human fibroblasts and instead causes such cells to undergo permanent growth arrest, also termed replicative senescence, characteristic of primary cells passed for multiple generations in culture. This senescence response appears to be dependent on the acute upregulation of p16(INK4A) induced by high levels of Ras expression, and it can be overcome by the loss of function of p16(INK4A) and p53 tumor suppressors (see Chapter 3), which may help to explain the selective pressure for inactivation of these genes in tumors containing Ras oncogenic mutations. 39,51
Not only is Ras itself mutated or overexpressed in cancer, but there are examples of Ras regulatory proteins, which can be affected as well. The best example is NF1, a Ras GAP mentioned earlier. Hereditary transmission of a defective NF1 allele predisposes an individual to a genetic disease called neurofibromatosis type 1 or von Recklinghausen’s neurofibromatosis. 52 Somatic mutations result in the inactivation of the second allele, leading to neoplastic development. Neurofibromatosis can manifest itself with the occurrence of multiple benign neurofibromas, as well as a high risk for malignancies of neural crest origin. In cells with defective NF1, cellular Ras accumulates in its GTP-bound state and thus is more active. 52 Other examples of Ras modulators involved in human hereditary diseases are the SH-2–containing tyrosine phosphatase SHP2 and the GNEF SOS1, whose mutations have been detected in patients affected by Noonan syndrome. 39,50
Signaling Downstream of RAS
Ras>Raf>Map Kinase Cascade
The most wellstudied effector of Ras is the serine/threonine kinase Raf (see Figure 2-1). Raf has been shown to bind to Ras and in many cases has been demonstrated to be indispensable for Ras functions such as cellular transformation. 53 In fact, activated Raf or v-Raf, a truncated form of c-Raf, was initially isolated as a retroviral oncogene. There are three known mammalian Raf isoforms designated A-, B-,and C-Raf (also known as Raf-1) (for review, see Ref. 54). C-Raf is ubiquitous in its tissue expression, whereas A-Raf and B-Raf expression is more restricted. Ras-mediated activation of Raf requires binding to two regions of this cytoplasmic kinase, both of which are located at the amino terminus. In the active state, Raf proteins form homo- or heterodimers, a process that is enhanced by Ras and inhibited by the extracellular signal regulated kinase (ERK) in a negative feedback loop. 55 Several phosphorylation events on both serine/threonine and tyrosine residues play a role in the full activation of Raf, 54,55 and there are major differences in certain phosphorylation sites between A/C-Raf and B-Raf. Also, B-Raf displays the highest basal kinase activity, which may explain why B-Raf, but not A/C-Raf, is frequently mutated in certain types of cancer. 55
Once activated, Raf can phosphorylate MEK (mitogen/extracellular-signal regulated kinase kinase), a dual-specificity kinase, on Ser218 and Ser222, leading to its activation (see review 56 and Figure 2-1). Partial activation can be seen by phosphorylation on only one serine. There are two isoforms of MEK, designated MEK1 and 2, both of which are expressed ubiquitously with an approximate sequence identity of 80%. MEK, once activated, can in turn activate mitogen-activated protein (MAP) kinase (also designated ERK). 56 Activation occurs via tandem phosphorylations on both threonine and tyrosine (Thr183-Glu-Tyr185) with the phosphorylation on tyrosine occurring first. There are two ERK isoforms (1 and 2), ubiquitously expressed and with very similar sequences. These proteins, 44 and 42 kDa, respectively, translocate to the nucleus, where they can activate a variety of proteins through phosphorylation on serine or threonine. For example, ERK can phosphorylate several of the members of the Ets family of transcription factors, and phosphorylation of Ets-1 by ERK dramatically increases c-fos transcription. ERK can also activate a variety of protein kinases via phosphorylation. For example, p90 RSK is a serine/threonine kinase, which has a role in protein translation and has been shown to be a substrate for ERK. 57
In addition to positive regulation of the MAP kinase pathway by phosphorylation, there are negative regulatory mechanisms that serve to attenuate activation of this cascade. A principal mode of this negative regulation is through a variety of phosphatases, a majority of which are dual specific, meaning they can dephosphorylate both serine/threonine and tyrosine residues. This is consistent with knowledge that ERK must be phosphorylated on both threonine and tyrosine to achieve maximal activation. 58
Functions of the MAP Kinase Pathway
As mentioned previously, the MAP kinase cascade mediates many Ras downstream functions (see Figure 2-1). ERK activation can lead to increased DNA synthesis and cell proliferation. In fact, activated forms of Ras, Raf, and MEK induce expression of cyclin D1, which plays a major role in early cell cycle progression. 59 Dominant negative mutants of members of this cascade can also block this induction in response to growth factor stimulation. In fact, cyclin D1 is one of the most frequently amplified genes in cancer, 60 and this locus is almost invariably translocated in certain lymphomas, associated with cyclin D1 overexpression. 59
Raf/Mek/MapK and Cancer
Davies and colleagues 61 identified B-Raf mutations in around 60% of human melanoma cell lines and primary tumors. Of note, these mutations, which in most cases involve the substitution of glutamate for valine 600 (V600E), were not consistent with mutations typically induced by UV. Lower frequencies of analogous mutations were observed in colon carcinoma and small-cell lung cancer (SCLC). 61 These mutations were further shown to oncogenically activate B-Raf, and recently mutant B-Raf has been successfully targeted in melanoma, as discussed later. In contrast to Ras and B-Raf, mutations of other Raf genes, MEK, or ERK are rare in cancer, although rearrangements causing aberrant activation of C-Raf have been recently reported in human prostate cancer. 62
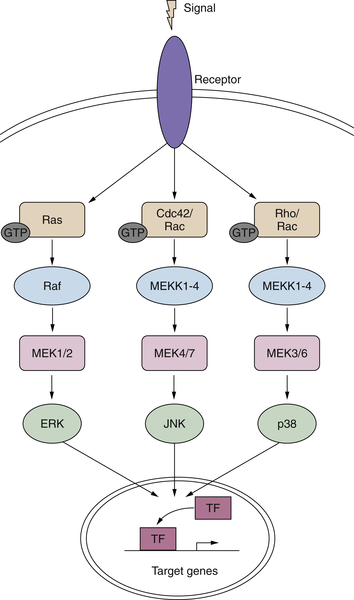
Figure 2-3 MAP kinase pathways in cancer Activation of the three MAP kinase cascades: ERK, JNK, and P38.
Other MAP Kinases
In addition to the ERKs, there are other MAP kinases belonging to distinct MAPK cascades with both different upstream activators and downstream effectors (Figure 2-3 ). The c-Jun N-terminal kinase (JNK)/stress-activated protein kinase (SAPK) and p38 MAP kinase have been demonstrated to modulate cellular responses to a wide variety of extracellular stimuli including mitogens, inflammatory cytokines, and UV irradiation (see review 63 ). There are three JNK genes, each with several alternatively spliced transcripts. In most cell types examined, including fibroblasts, epithelial cells, and neuronal-like PC12 cells, sustained activation of JNK/SAPK has been reported to induce programmed cell death, whereas transient activation can promote cell survival. 63 There is evidence for some redundancy among these three genes, as each of the single knockouts as well as the JNK1/JNK3–/– and JNK2/JNK3–/– double knockouts is viable, but mice lacking both JNK1 and JNK2 are embryonic lethal. However, it has been demonstrated that the three JNK isoforms can also exhibit distinct properties, for example, in their capacity to interact with the oncogene Jun. 63
In contrast to its ability to activate the MAPK/ERK cascade, H-ras only minimally perturbs JNK/SAPK. However, overexpression of the constitutively activated mutants of the small G-proteins, Rac and Cdc42, leads to the robust stimulation of JNK/SAPK activity. The pathways leading to JNK activation mirror those seen for ERK. Thus, a variety of MAP kinase kinases (MKK) can phosphorylate the various JNK isoforms. 56,63
As with the ERKs, JNK activation results in phosphorylation of certain transcription factors and increases their transcriptional activity at promoters containing response elements for these factors. 63 Some of the transcription factors activated by ERK or JNK were initially discovered as retroviral oncogenes in mice and chickens, respectively. The FBJ and FBR murine viruses contain the Fos sequence under the viral LTR promoter and exhibit changes in regulatory phosphorylation sites that make them more active than the proto-oncogene, 64 and Jun was identified as an avian retrovirus. 65 Overexpression of Fos can cause transformation of cells as well. 66 Jun and Fos belong to the AP-1 family of transcription factors. In response to various stimuli, including UV irradiation, environmental stresses, and PKC activation, Jun homodimers and Jun-Fos heterodimers bind to AP-1 target sequences such as 12-O-tetradecanoylphorbol-13-acetate (TPA) responsive elements. 67
C-Myc
The Myc family includes four transcription factors, c-Myc, N-Myc, L-Myc, and S-Myc, involved in the control of cell growth, differentiation, and apoptosis. 68,69 Myc proteins form heterodimers with another transcription factor, Max, through a basic-region/helix-loop-helix/leucine-zipper domain and bind a specific DNA consensus sequence called E-box to activate the transcription of target genes. In the absence of Myc, Max forms a complex with Mad/Mnt proteins and acts as a repressor of Myc transcriptional targets. A large number of genes are regulated by Myc, including p21(CIP1), cyclin D1/D2, E2F2, and the microRNA miR-17-92 cluster. Because the E-box sequence is very frequent in the human genome, more sophisticated approaches, such as genome-wide chromatin immunoprecipitation, have been used to identify authentic and physiologically relevant Myc target genes. 69 Enhanced expression of Myc proteins is estimated to occur in up to 70% of human malignancies. However, the deregulated expression of these proteins is not sufficient to induce cell transformation, implying that additional genetic events are required. One such event is the activation of the Ras pathway, which affects Myc factors at different levels, including posttranslational stabilization and inhibition of the antagonizing transcription factors FOXO. Also, because high levels of Myc can trigger apoptosis, tumor cells may need for their survival concomitant oncogenic events, such as Ras activation or loss of ARF or p53 tumor suppressors. 68,69 In lymphoid cancers, c-Myc is often found in translocations adjacent to a strong promoter, such as that of the immunoglobulin genes. In other cancers such as breast and lung carcinoma, the genomic locus encoding c-Myc is amplified. c-Myc overexpression can also result from the aberrant activation of upstream pathways, such as in the case of Wnt signaling discussed later. 68,69 Gene amplification is also the mechanism responsible for the increased expression of N-Myc commonly observed in certain cancers, including retinoblastoma, glioblastoma, and medulloblastoma. Of note, N-Myc overexpression in neuroblastoma strongly correlates with an advanced clinical stage, and it is taken into consideration for the assessment of the treatment of these malignancies. 70
Oncogenes and Survival Signaling
The regulation of cell survival and cell death is of extreme importance both in the development of an organism and in the physiologic functions of the adult. During development of a multicellular organism, certain cells are eliminated by a process known as apoptosis or programmed cell death and others are permitted to survive. The deregulation of these processes can lead to a variety of malformations resulting in deformities or, in extreme cases, incompatibility with life. In adulthood, regulation of cell survival is equally important for proper homeostasis. Damaged cells must be removed, and terminally differentiated cells must be sustained. If this fails to occur, mutations leading to cancer may accumulate. Pro-apoptotic and anti-apoptotic proteins regulate these processes, and many of the oncogenes already discussed modulate cell survival in a positive fashion. Thus, oncogenes can influence proliferation, cell survival, or both, contributing to cellular transformation in a cooperative fashion.
The Bcl-2 Family
The Bcl-2 family of proteins consists of more than 15 members, which can be subdivided into three classes based on functions and the number of Bcl-2 homology (BH) domains present. 71,72 The anti-apoptotic members, including Bcl-2, Mcl-1, and Bcl-XL, contain four BH domains (BH1 to BH4). The pro-apoptotic BCL-2 factors are divided into the effector proteins, such as BAX and BAK, which also contain four BH domains, and the “BH3 only” pro-apoptotic proteins, such as BID and BIM, which include only the BH3 domain. Proteins in all three classes have the ability to form either homo- or heterodimers with one another and play distinct roles in regulating mitochondrial membrane permeabilization 71,72 (Figure 2-4 ).
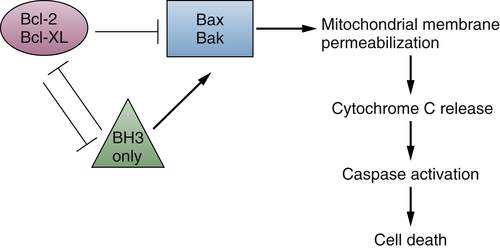
Figure 2-4 Bcl-2 family member interactions regulate cell death
The involvement of Bcl-2 and cancer has been firmly established. Not only was the gene cloned as a translocation from a lymphoid tumor, but mice expressing a Bcl-2-immunoglobin “mini-gene” that mimicked the translocation seen in human cancers showed follicular hyperplasia that progressed to lymphoma. The Bcl-2 genomic locus has been found to be translocated in several tumor types, including follicular lymphomas and chronic lymphocytic leukemia, and other oncogenes, such as Ras, can stimulate Bcl-2 expression. 72 The Mcl-1 and Bcl-X genes are frequently amplified in tumors, 60 and overexpression of anti-apoptotic family members as well as downregulation or inactivation of pro-apoptotic proteins has been observed in several human cancers. 72
PI3K-Dependent Pathways
PI3K is a lipid kinase that catalyzes the transfer of the γ-phosphate from ATP to the D3 position of phosphoinositide (PtdIns), generating PtdIns3P, PtdIns(3,4)P2, and PtdIns(3,4,5)P373 (see Figure 2-1). These lipids can act in a variety of cascades. PI3K activation has been demonstrated to play an important role in cell survival signaling in a number of cell types. There are three classes of PI3Ks, which exhibit variability with respect to their method of activation or their preferred lipid substrate.
The class I of PI3K, and in particular the subclass IA, is the most relevant for cancer and the best characterized. It consists of two subunits encoded by two distinct loci, a regulatory and a catalytic subunit. 37 The regulatory subunit is a 50- to 85-kDa protein that is tightly associated with the p110 catalytic subunit. The classical mode of PI3K activation involves its binding to the phosphorylated tyrosine residues of receptor tyrosine kinases via the two SH2 domains of p85. This results in the recruitment of the p85-p110 heterodimer to its substrate at the cell membrane and in a conformational change that relieves the basal inhibition of p110 catalytic activity. 37 In addition, it has been demonstrated that PI3K can be activated independently of receptor binding by the small G-protein Ras 37,74 (see Figure 2-1). Furthermore, the γ isoform of PI3K is activated by heterotrimeric G-proteins. 37 Thus it is clear that PI3K can be activated in response to a wide variety of upstream signals.
There are several known downstream effectors of PI3K. These include Rac, p70(s6k), certain isoforms of PKC, and, most relevant to the discussion of cell survival, Akt/PKB. 37,73 Akt has been shown to be responsible for PI3K-dependent cell survival signaling and is the cellular homologue of the viral oncogene v-Akt. The three human homologues identified encode 57-kDa serine/threonine kinases that contain an amino-terminal pleckstrin homology domain, which binds to the activated PtdIns products of PI3K. These lipids are believed to mediate the localization of this cytoplasmic protein to the plasma membrane. In addition, phosphorylation of Akt on two residues, a serine and a threonine, is required for full activation. These events are catalyzed by two different kinases, one of which, PDK1 (PtdIns(3,4,5)P3 dependent kinase) specifically phosphorylates Thr308; the other, the mTOR (mammalian target of rapamycin) complex 2, phosphorylates Ser473. 37,74 Once activated, Akt directly phosphorylates different proteins, such as BAD, forkhead box O transcription factors, glycogen synthase kinase 3 (GSK3), and tuberous sclerosis 2 (mTOR signaling), thus regulating various processes, including cell survival, proliferation, and protein synthesis 74 (see Figure 2-1).
The striking anti-apoptotic effect of both PI3K and its downstream effector Akt, as well as the fact that these two genes were initially found as transforming viral oncogenes, suggested that these two genes might also be involved in human cancer. Indeed, a myristoylated constitutively active PI3K can cause cellular transformation in chicken embryo fibroblasts. 75 The gene encoding the p110α catalytic subunit is frequently mutated in certain common epithelial cancers, such as breast, colon, and prostate. One class of mutation, which promotes constitutive signaling, involves the kinase domain near the activation loop. The second class of p110α mutations is predicted to abrogate the inhibitory intermolecular interaction between p85 and p110. Consistent with this model, mutations affecting the p85 subunit have recently been identified in glioblastomas and in colon and ovarian cancers. 74 Activating mutations of the three Akt isoforms have been also described, although they appear to be far less common than those involving the p110α locus. In addition, certain cancers can harbor amplifications of p110α or Akt subunits. 74
Further evidence of the involvement of the PI3K/Akt pathway in cancer stems from the discovery of PTEN, one of the most frequently inactivated tumor suppressor genes in different types of tumors, including glial and endometrial tumors, as well as melanoma, prostate, renal, and small-cell lung carcinomas (for a review, see Ref. 76). Germline mutations at the PTEN locus cause inherited cancer syndromes such as Cowden disease, Lhermitte-Duclos disease, and Bannayan-Zonana syndrome. PTEN dephosphorylates the 3 position of phosphatidylinositol, thus directly opposing PI3K activity 76 (see Figure 2-1).
Cytokine Receptor Signaling
A large number of cytokines, hormones, and growth factors have been shown to activate a class of receptors that lack significant sequence similarity to the RTKs and are grouped under the definition of cytokine receptors. 77 They share a common structural motif in their extracellular domains, including conserved cysteine residues, and lack intrinsic enzymatic activity. The cytokine receptors either homodimerize on ligand binding (receptors for growth hormone, prolactin, erythropoietin, and thrombopoietin) or are composed of two distinct subunits that heterodimerize in response to ligand interaction. This latter group of receptors is composed of a ligand-specific chain and a common chain shared by different cytokines. This includes the receptors for interleukin-6 (IL-6), IL-11, oncostatin M, LIF, cardiotrophin-1, and ciliary neurotrophic factors, all sharing a common chain called gp130; the receptors for IL-3, IL-5, and GM-CSF that share the β common chain; and the receptors for IL-2, IL-4, IL-7, IL-9, and IL-15 that share the γ common chain. 78
The Janus kinases (JAKs) originally identified as signaling molecules in the interferon pathway are essential transducers of the signal originating from cytokine receptors. 79 Four mammalian JAKS have been identified, JAK-1, JAK-2, JAK-3, and Tyk-2. These kinases are associated with the receptors and contain both a catalytic domain and a pseudokinase domain that is involved in the inhibition of JAK activity in the absence of the ligands. On ligand binding, the activated JAKs cause phosphorylation of the receptor and of molecules containing either a phosphotyrosine-binding or an SH2 domain. These molecules comprise the signal transducers and activators of transcription (STATs).
Seven mammalian STATs have been identified (STAT-1, STAT-2, STAT-3, STAT-4, STAT5a, STAT5b, and STAT-6), and differential splicing increases the number of these molecules. In addition to the SH2 domain, their structure is characterized by a DNA-binding domain and several protein-protein interaction domains. After becoming phosphorylated on tyrosine, STATs form homo- or heterodimers through their SH2 domain and translocate to the nucleus, where they activate target genes (Figure 2-5 80,81 ). In addition to the cytokine receptors, the JAK/STAT pathway has been shown to transduce signals from a number of tyrosine kinase receptors, including those for EGF, PDGF, and CSF-1. 80
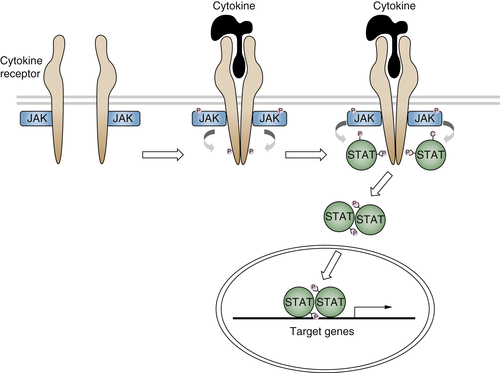
Figure 2-5 Cytokine receptor signaling The binding of cytokines to their receptors activates the JAK/STAT pathway through a series of phosphorylations.
Several components of the cytokine receptor signaling pathways have been implicated in uncontrolled cell proliferation and cancer. The myeloproliferative virus (MPLV), an acute transforming retrovirus, contains an oncogene called v-mpl, which is a truncated version of a member of the hematopoietin receptor family c-mpl whose ligand has been identified as the thrombopoietin. Ectopic expression of c-mpl induces a lethal myeloproliferative disease in mice, 82 and activating mutations have been identified in myelofibrosis with myeloid metaplasia in humans. 83 The first evidence of involvement of the JAKs/STATs in naturally occurring cancer was the finding of an activating mutation in the Drosophila Hop kinase, a member of the JAK family, that caused a leukemia-like phenotype. 84 Stronger evidence implicating the JAKs in a human cancer came from the identification of a chromosomal translocation in a human leukemia resulting in the constitutively activated fusion protein TEL-JAK2. 85 In 2005, various groups identified a mutation in the JAK2 gene in a large proportion of patients with different types of myeloproliferative neoplasms, including polycythemia vera (PV) and essential thrombocythemia. This mutation involves the substitution of a phenylalanine for valine 617 (V617F) and is believed to disrupt the autoinhibitory activity of the pseudokinase domain, resulting in cytokine hypersensitivity and cytokine-independent growth. 81 It has been shown that in hematopoietic cells containing JAK2(V617F), signaling pathways involved in cell proliferation and survival are activated, including STAT, MAPK, and PI3K-AKT. 81 More recently, activating mutations in JAK2 exon 12, which encodes a region near the pseudokinase domain, were identified in JAK2(V617F) negative PV patients. 81 Although activating mutations of STAT3 have been identified only in a particular type of liver tumor, 86 this transcription factor is very frequently activated through various mechanisms in cancer cells. It is thought not only to participate in tumor cell proliferation, survival, and invasion, but also to suppress antitumor immunity and to maintain a procarcinogenic inflammatory microenvironment. 87
Neurotransmitters
The transmission of signals generated by the reception of chemical and physical stimuli from the external and internal environments is mediated by a large variety of small molecules known as neurotransmitters. These molecules include acetylcholine; amino acid derivatives such as epinephrine, norepinephrine, serotonin, and dopamine; and peptides such as the angiotensins, β-endorphin, enkephalins, and somatostatin. These ligands can trigger two types of receptors: ion-channel–linked receptors or receptors with seven membrane-spanning domains, which interact with heterotrimeric G proteins composed of α, β, and γ subunits. After binding to their specific ligand, the G-protein coupled receptors (GPCRs) undergo a conformational change, which results in a switch from the inactive GDP-bound Gα to an active GTP-bound state and the dissociation of the Gβγ subunits. Different subfamilies of Gα proteins exist that activate various signaling pathways. For example, Gαs and Gαi, respectively, stimulate or inhibit adenylyl cyclase, provoking an increase (or a decrease) of cyclic AMP levels, which can then activate the protein kinase A (Figure 2-6 ). The members of another Gα subfamily, Gαq, activate PLC-β, which catalyzes the cleavage of phosphatidylinositol biphosphate into DAG and IP3. DAG then stimulates PKC, whereas IP3 mobilizes the intracellular stocks of calcium (see Figure 2-6). The Gβγ subunits are also implicated in the signaling cascade, by regulating the activity of different effectors, such as phospholipases, ion channels, and various kinases. Of note, GPCR activation can impinge on other transduction pathways, including Rho and Ras GTPases or MAP kinases, while the mechanisms involved are not completely elucidated (for review, see Ref. 88).
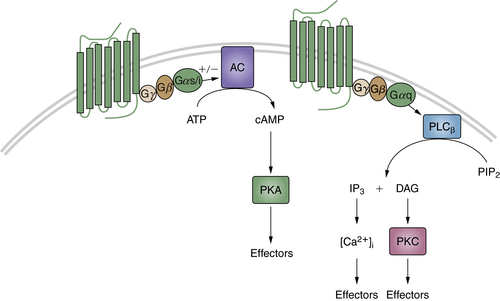
Figure 2-6 G-protein coupled receptors (GPCRs) Diagram showing cyclic adenosine monophosphate (cAMP) and phospholipase C-β (PLCβ) transduction pathways activated by GPCRs.
The ability of GPCRs to activate various transduction pathways that regulate cell differentiation and proliferation strongly suggests a potential role of these receptors in tumorigenesis. Indeed, activating mutations of the thyroid-stimulating hormone receptor commonly occur in thyroid adenomas and carcinomas, and germline mutations cause familial non-autoimmune hyperthyroidism. 88 Another example is illustrated by studies on two distinct groups within a subset of growth hormone–secreting human pituitary tumors. 89 In one group, Gαs was found to be constitutively active, resulting in elevated adenylate cyclase activity and growth hormone levels. This activation was due to point mutations either in a site at which cholera toxin inactivates Gαs [Arg 201 → Cys/His] or at a residue equivalent to a GTPase-inhibiting mutation that causes malignant activation of Ras p21 [Gln 227 → Arg]. Because both mutations have the effect of destroying GTPase activity, Gαs [designated gsp] becomes constitutively activated in a manner analogous to the oncogenic activity of Ras p21. The two mutations are located in regions that are highly conserved among Gα proteins isolated from diverse eukaryotic species, and activating mutations have been identified in some human adrenal, pituitary, and other endocrine tumors. 90 Although mutations in GPCRs and G proteins have been identified in some tumors, the most common mechanisms of GPCR activation in cancer cells are receptor overexpression and autocrine stimulation, 88 as has been shown, for example, for the gastrin-releasing peptide, 91 angiotensin II, 92 and cholecystokinin 93 in pancreas and prostate cancers. Moreover, the receptors for certain neuropeptides and/or neurotransmitters, including acetylcholine, bradykinin, bombesin, and endothelin, can promote the proliferation and invasion of different types of tumor cells through cross talk with the EGFR or the insulin-like growth factor 1 receptor. 94 Of note, the specific expression of neurotransmitters and their cognate receptors in tumors arising from the endocrine system make this signaling a promising target for cancer diagnosis and therapy. 95,96 Finally, it is worth mentioning that other families of ligands triggering GPCRs play important roles in tumor initiation and progression. This is the case, for example, with the prostaglandins, which mediate chronic inflammation, increasing the risk of tumors, and the chemokines, crucially involved in cancer metastasis. 88,94
Wnt Signaling
Wnts comprise a highly conserved multimember ligand family and play important roles in a variety of developmental processes, including patterning and cell fate determination. 97,98 In different adult tissues, Wnts also play important roles in stem/progenitor cell maintenance and differention. 98–100 Wnts bind to two co-receptors, the seven-transmembrane Frizzled and single-membrane–spanning low-density receptor-related protein 5/6 (LRP5/6), resulting in the phosphorylation of the LRP5/6 intracellular domain. 101 The phosphorylated LRP5/6 provokes the inhibition of a multiprotein complex, referred to as the destruction complex, which includes axin, GSK3, casein kinase 1 (CK1), and adenomatous polyposis coli (APC). In the absence of Wnt ligands, this complex induces the sequential phosphorylation of β-catenin by CK1 and GSK3, which provokes its degradation through the ubiquitination pathway. Thus, Wnt-induced inhibition of the destruction complex blocks β-catenin degradation and causes its accumulation in the cytoplasm in an uncomplexed form. The latter is then translocated to the nucleus, where it binds to transcription factors belonging to the T-cell factor/lymphoid enhancer factor family and activates the expression of Wnt target genes, including the proto-oncogene c-myc and cyclin D1 (Figure 2-7 100 ).
The prototype Wnt gene was originally identified as a cellular gene activated by integration of the mouse mammary tumor virus. 102 Later studies indicated that targeted expression of certain Wnts in transgenic mice caused mammary gland hyperplasia, and several Wnt genes exhibit the ability to transform various epithelial 103 and fibroblast murine cell lines. 104 Recent evidence indicates that Wnt signaling is constitutively activated through an autocrine mechanism in different types of human cancers, including breast cancer, 105 non–small-cell lung cancer, 106 and sarcoma. 107 More commonly, specific downstream components of the Wnt pathway have been implicated in human cancers. Genetic alterations of β-catenin have been identified in human tumors and cancer cell lines, including colon cancer, melanomas, and hepatocellular carcinomas. 97,108 These mutations affect the sites of phosphorylation of β-catenin and result in the inhibition of its degradation, leading to the stabilization of the protein in the cytosolic and/or nuclear compartments.
The APC tumor suppressor gene product, which is required for β-catenin phosphorylation and degradation, also regulates the amount of cytosolic β-catenin. Germline mutations in the APC gene are responsible for familial adenomatous polyposis (FAP), a dominantly inherited syndrome characterized by the formation of hundreds of colorectal adenomas, some of which inevitably progress to colorectal cancer. Inactivation of the APC gene leading to increased cytosolic β-catenin is also found in 80% of sporadic colon cancers, and it represents an early event in tumor progression. 97,108 The major initiating event in the remaining 20% of colon cancer involves mutations in the β-catenin gene 97,108 or, as recently uncovered by next-generation sequencing, fusions of the R-spondin genes, a family of secreted proteins that enhance Wnt signaling. 109 Of note, these events occur only in those cancer cells with intact APC, implying that activation of this signaling pathway affects almost all colon cancers.
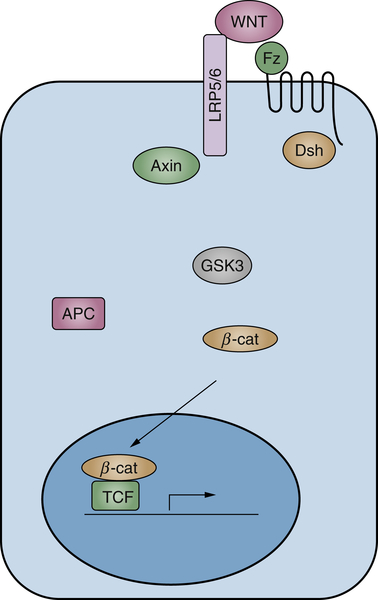
Figure 2-7 Diagram showing the major known components of Wnt signaling in cancer Simplified scheme of canonical Wnt signaling.
Hedgehog/Patched Signaling
The Hedgehog/Patched signaling pathway was first identified in Drosophila, where it plays an important role in a number of developmental processes, including cell fate determination and patterning. 110,111 Although the major core components of the pathway are conserved among species, many differences exist between the fly and vertebrate Hedgehog signaling. In vertebrates, in the absence of the Hedgehog ligands, Sonic, Indian, and Desert Hedgehog, their receptor Patched prevents, through an unknown mechanism, the translocation of the seven-transmembrane domain protein Smoothened to the primary cilium. Under such conditions, the full-length Gli transcription factors bind to suppressor of fused (Sufu) and, following multiple phosphorylation, are cleaved into repressor forms of Gli that inhibit the transcription of Hedgehog target genes. Hedgehog binding to Patched relieves the inhibition of Smoothened, which can then translocate to the primary cilium, where it promotes the disassembly of the Sufu-Gli complex. Gli proteins are shuttled into the nucleus, where they become transcriptionally active, probably after additional posttranslational modifications 112,113 (Figure 2-8 ).
Related posts:




Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


