31.1
Introduction
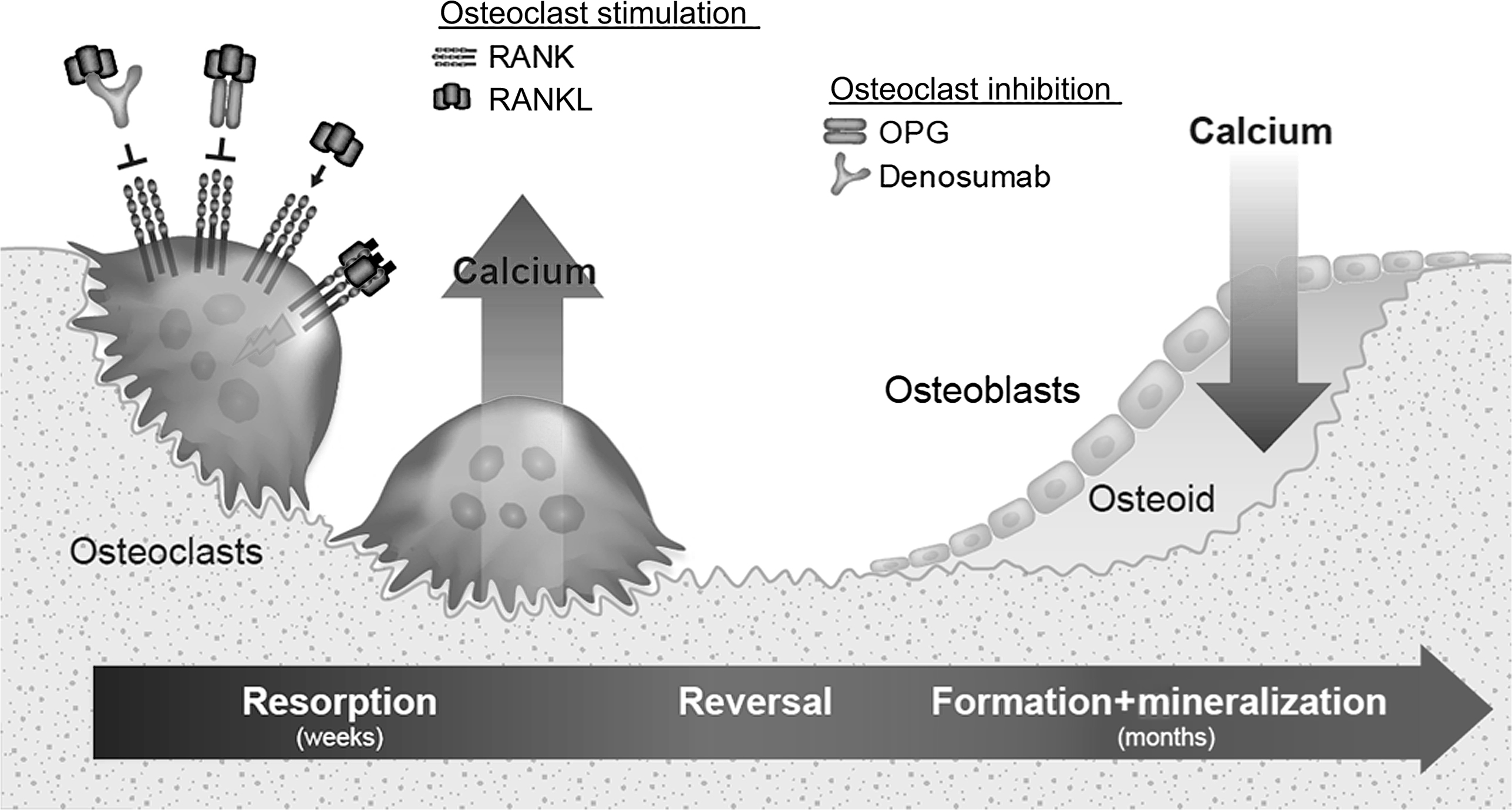
Dobzhansky’s quote, “Nothing in biology makes sense except in the light of evolution” provides a unique framework for contemplating the origins and contemporary roles of bone remodeling. There are several forms of bone remodeling, and one form that has long puzzled scientists in terms of its purpose is the continuous lifelong turnover of mineralized bone matrix by basic multicellular units (BMUs). In BMU remodeling, osteoclasts resorb a discrete packet of bone, and osteoblasts later refill the resorption space with collagenous matrix that gradually mineralizes with calcium and phosphate ( Fig. 31.1 ). This remodeling plays important roles during skeletal development and growth, including establishment of marrow cavities and conversion of primary or woven bone into stronger lamellar bone. The absence of remodeling during growth leads to osteopetrosis . Bone modeling , which is most active during early life stages, describes processes by which osteoclasts and osteoblasts work in a spatially independent manner to establish the shape and general architecture of the skeleton. Modeling optimizes bone geometry over long periods of growth and habitual loading, whereas lifelong BMU remodeling generally maintains this optimized structure by resorbing and replacing bone at the same microlevel location .
In contrast to other forms of remodeling a unique attribute of continuous BMU remodeling in the mature skeleton is that it replaces mineralized lamellar bone with the same type of tissue. In recognizing this feature, Parfitt noted, “There is probably no physiological function other than bone remodeling which has attracted so much study in the face of so much uncertainty about why it occurs” . The osteoporosis field is indeed armed with an intimate understanding of how bone remodels, while debating whether its primary role serves metabolic or biomechanical imperatives, or both. The metabolic role of BMU remodeling typically centers on calcium homeostasis, while the biomechanical value of remodeling is generally predicated on the notion that bone matrix eventually becomes unfit for load-bearing and thus requires continuous removal and replacement.
Bone remodeling can be experimentally modified in humans and laboratory animals to study the effects on bone strength and mineral homeostasis, but this approach might not answer the question of why continuous BMU remodeling evolved in the first place. According to Eaton, it has been 20,000 years or more since the human gene pool consistently interacted with the bioenvironmental circumstances typical of those for which our genome was originally selected . This point is particularly relevant to this chapter’s discussions on fracture risk, which in the current era tends to focus on factors that influence the risk of fragility fractures. In contrast, fractures in the natural world generally have unpredictable and traumatic etiologies, such that any reduction in bone strength had the potential to increase fracture risk in our ancient forebears.
Animal studies can provide important insights into remodeling’s contemporary roles, and it is not uncommon for laboratory animals treated with remodeling inhibitors to exhibit modest declines in blood calcium and increased bone mass and strength. However, such changes will not meaningfully alter a lab animal’s prospects of survival or reproductive success, which are the key evolutionary drivers. An animal’s life in the natural world is often nasty, brutish, and short, and natural selection can work efficiently in that state, acting most powerfully on traits that govern short-term survival and reproduction.
An exclusive focus on evolutionary biology is also unlikely to yield clear answers to why BMU remodeling evolved, in part because evolutionary theories are often difficult to reduce to falsifiable hypotheses. Furthermore, the purpose of an original trait can change over time; while feathers are a trait birds rely on for flight, feathers were originally used by flightless terrestrial dinosaurs for warmth, mating displays or camouflage .
Comparative biology is another discipline that can provide some clues regarding evolutionary origins, through examination of the phylogenetic origins of a trait, along with the environmental circumstances that might have allowed the trait to confer survival advantages and/or differential reproductive success.
This chapter attempts to bridge evolutionary theory, contemporary experimental data, and comparative biology in considering why the bones of skeletally mature vertebrates continuously remodel throughout life. In deliberating why a given trait exists, it is essential to consider all of the traits benefits and costs at the level of the entire host . The reasons bone continuously remodels need not reside within the skeleton, or at least not entirely. In addition, as with any trait, BMU remodeling need not provide perfect solutions for any of the functions it serves. Evolution by natural selection is an incremental, stepwise process, beset by many constraints set in place by phylogenetic heritage, previous developmental pathways, and general body architecture ( bauplan ). These constraints can lead to suboptimal traits that are still sufficiently beneficial to accumulate within a population . This evolutionary principle would allow, for example, that bone remodeling in the interest of calcium homeostasis could weaken bone, even to the point of fracture, while still having a net positive effect on survival and reproductive success that would secure its natural selection.
The earliest evidence for a BMU remodeling-like activity, which may have provided a template for the continuous remodeling trait, might be found in the tooth-bearing bones of our ancient vertebrate ancestors, the bony fish (see Section 31.9 ). There, osteoclasts resorb discrete packets of mineralized bone matrix to allow teeth to erupt and to move with the perpetual growth of fish jawbones. Tooth attachment in fish and other animals is maintained via the refilling of resorption spaces in alveolar bone, and this refilling is a hallmark of BMU remodeling and a point of emphasis in this chapter. While remodeling-like activity is evident in fish jaws, it is conspicuously absent from their postcranial skeletons. Early descendants of fish (i.e., primitive tetrapods) may have coopted ancestral remodeling traits originally used to secure dentition and applied it throughout their skeletons. Possible drivers for this adaptation include the newfound need for terrestrial animals to access skeletal calcium reserves, to lighten underloaded regions of their skeletons to minimize energy expenditure, and/or to renew bone matrix that might be damaged by greater skeletal loading due to the effects of gravity on terra firma. The singular trait of BMU remodeling can manage these and other important biological imperatives. We cannot know which of these features led to swift adoption of the remodeling trait by terrestrial vertebrates and aquatic mammals, but the multidisciplinary approaches laid out in this chapter suggest that calcium homeostasis is remodeling’s original and still central role.
31.2
The basics of bone remodeling
The broadest definition of bone remodeling is the replacement of one packet or type of bony tissue with another . The replacement of mineralized bone begins with its resorption, accomplished by various specialized cells capable of acidifying and resorbing mineral crystals and destroying the associated collagenous matrix through proteolysis. Osteoclasts and chondroclasts, which resorb mineralized bone and cartilage, respectively, probably existed 600,000,000 years ago . Odontoclasts and cementoclasts capable of resorbing tooth structures probably have earlier origins and may have been preceded by poorly characterized scleroclasts that mobilize the abundant calcium present in fish scales .
The capacity to resorb mineralized tissue may have originated nearly 1 billion years ago , but the more ancient resorptive processes were rarely followed by replacement with the same type of mineralized tissue. For example, most of the bone resorbed for tooth eruption is replaced (spatially) by tooth structure rather than by new bone . The resorption of growth plate cartilage is followed by replacement with mineralized bone matrix more suitable for load bearing. And rapidly deposited primary , chondroid , fusiform , woven, or plexiform bone can be remodeled into lamellar bone possessing superior material strength properties, thereby enhancing a bone’s strength relative to its mass. These and other forms of bone remodeling, where one type of skeletal tissue is replaced by another, have accepted utilities that provoke little debate. The rest of this chapter will therefore focus on continuous BMU remodeling, wherein mineralized bone matrix is removed and replaced by similar tissue.
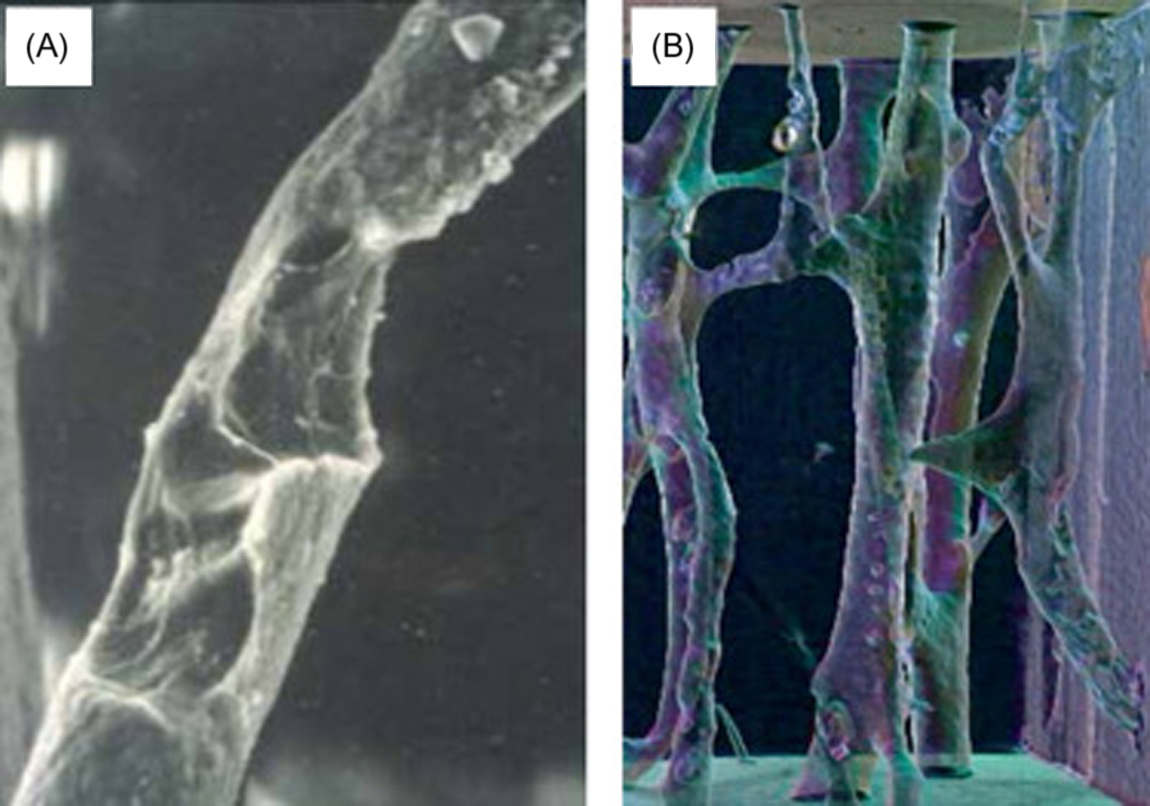
The mechanics of BMU remodeling, described in rich detail by Parfitt (see also Chapter 36 : Bone mineral acquisition in utero and during infancy and childhood), will be greatly simplified here. BMU remodeling begins with osteoclastic bone resorption, which invariably mobilizes skeletal calcium into the blood ( Fig. 31.1 ). When acting upon trabecular and endocortical surfaces, osteoclasts engaged in remodeling create relatively deep resorption spaces with jagged scalloped bases, known as Howship’s lacunae. Acting within cortical bone, osteoclasts dig long tunnels that primarily run parallel to the bone’s major axis ( Fig. 31.2 ), creating new porosity that is mostly refilled by osteoblasts to form a new osteon (i.e., Haversian system) that contains its own new blood and nerve supply. Thus endosteal (trabecular and endocortical) remodeling is primarily a surface-based phenomenon, while intracortical (Haversian) remodeling is primarily a tunneling phenomenon.
Molecular pathways regulating osteoclasts and the initiation of remodeling have been reviewed previously . One such pathway was uncovered in the 1990s with the discovery of osteoprotegerin (OPG), a tumor necrosis factor (TNF) receptor superfamily member discovered by its ability to inhibit osteoclasts when overexpressed in transgenic mice . OPG acts by binding to a TNF superfamily member called RANKL (receptor activator of nuclear factor kappa-B ligand) ( Fig. 31.1 ). RANKL activates a receptor called RANK that is found on osteoclasts and their precursors , thereby promoting osteoclast formation, activation, and survival. When RANKL binds to OPG, it cannot activate RANK and bone resorption quickly subsides . RANKL and OPG are among many factors regulating osteoclasts and are highlighted here because of their dominant effects on osteoclasts and because of their utility as research tools for studying the consequences and potential roles of BMU remodeling (the focus of Section 31.7 ).
As osteoclasts complete the resorptive phase of BMU remodeling the resorption spaces are refilled with organic collagenous bone matrix called osteoid ( Fig. 31.1 ). Osteoid is deposited in resorption spaces at about 1 μm/day, much slower than the 50 μm/day rate at which osteoclasts can resorb bone . Extracellular calcium, including blood calcium, is taken up by osteoid as it undergoes a rapid (primary) mineralization phase during its initial deposition, which is followed by a slower secondary phase of mineralization during which the matrix attains its full degree of strength and stiffness . Estimates on the full duration of a remodeling cycle in humans, including secondary mineralization, range from 7 to 9 months to as long as 1–5 years , though newly remodeled bone probably attains significant load-bearing capacity much sooner . The favorable safety and efficacy profile of antiresorptive therapies is based in part on the fact that the refilling of existing resorption spaces continues when those agents inhibit active osteoclasts and reduce remodeling activation .
One unfortunate attribute of BMU remodeling, from a strictly biomechanical perspective, is its propensity to under-fill resorption spaces, particularly with advancing age and with estrogen deficiency . In this “negative bone balance” state, bone remodeling progressively decreases bone mass in a manner that can increase fracture risk, particularly in postmenopausal women. There are, however, important settings where a negative bone balance benefits a host. For example, unbalanced remodeling can lead to the elimination of underloaded bone , which minimizes energy expenditure for locomotion (see Section 31.4 ). Repositioning of teeth to maintain their occlusion, to allow their eruption, and to accommodate jawbone growth also requires that the resorption of alveolar bone at the tooth’s leading edge exceeds the subsequent refilling . Bone balance can be modulated by varying the depth of resorption and/or the degree, duration, and lag time of refilling, thereby allowing the basic remodeling trait to accomplish a variety of tasks unrelated to the maintenance of bone strength.
Whether balanced or imbalanced, a key feature of BMU remodeling is the “remodeling transient,” which describes the temporary deficit of bone volume and mineral that necessarily exists between the rapid creation of resorption spaces and their slower refilling with mineralizing osteoid . During this interval a remodeled region of the skeleton contributes positively to the extracellular calcium pool while facing its own temporary volume deficit that manifests as lost structure and strength. In this manner, BMU remodeling provides a “loan” of extracellular calcium that may be used for metabolic needs and which is repaid when the resorption space is refilled and mineralizes. An increase in the rate of remodeling will provide more extracellular calcium at the expense of skeletal structure. Here is a key example wherein two biological imperatives, calcium homeostasis and bone strength, conflict in a manner that is mediated by the singular pleiotropic trait of BMU remodeling.
When bone mass declines for any reason, including those that confer metabolic benefits to its host, any resulting decrease in bone strength is an unintended but unavoidable consequence. Through examples described later, one might appreciate that BMU remodeling is nature’s way of minimizing decreases in bone strength that occur when osteoclasts serve metabolic imperatives. With the BMU remodeling trait at our disposal, fracture risk may be minimized even when BMU remodeling activity leads to absolute reductions in bone strength. Such outcomes can indeed coexist if the absence of the BMU remodeling trait would result in even greater reductions in bone strength when the skeleton is called upon to serve metabolic imperatives. This hypothetical outcome is predicated on evidence that osteoclastic bone resorption can occur without the refilling of resorption spaces, as is the case with modeling-based resorption drifts , and on evidence that vertebrates lacking the continuous remodeling trait still resorb bone matrix when extracellular calcium demands are high (see Section 31.9 ). Thus osteoclasts can be expected to catabolize the skeleton to serve important metabolic needs whether or not the bone strength-sparing features of the remodeling trait are available. There are at least two key features unique to BMU remodeling that minimize strength losses even as bone mass is reduced. One is its “smoothing” function, whereby deep jagged resorption spaces are at least partially refilled with a smooth covering of osteoid that can rapidly improve stress distribution, and its “placeholder” function, whereby the refilling of resorption spaces helps maintain original bone architecture that is optimized by the modeling apparatus over long periods of growth and habitual loading .
31.3
Skeletal involvement in managing metabolic imperatives: roles and consequences
Fractures frequently compromise survival and reproduction in the natural world, yet evolution seems to have programed skeletons to sacrifice mass and strength when called upon to serve more pressing metabolic needs. For example, underloaded bone can be “remodeled away” to lighten the skeleton, which can lessen energy expenditure at the cost of reducing skeletal strength ( Section 31.4 ). And faced with inadequate dietary calcium, or a decreased ability to absorb or retain calcium, a host can increase bone remodeling to mobilize more skeletal calcium, which will reduce bone mass and strength even to the point of enabling fragility fractures ( Section 31.5 ). Other sources of evidence suggest that avoiding fractures is not an overriding priority of the host or its skeleton. For example, bone mass, density, and strength in rodents, monkeys, and humans tend to begin an inexorable decline before midlife, well before reproductive senescence . The number of causes of secondary osteoporosis has been described as vast , which suggests at a molecular level that maintaining bone strength is not the host’s prime directive. It is perhaps no coincidence, then, that fracture healing is one of the most robust tissue repair processes in nature; the evolutionary selection and conservation of such an effective repair trait suggest that a substantial number of terrestrial vertebrates fractured and then healed to the point of being able to procreate and pass along their healing traits.
There are many examples where BMU remodeling reduces bone strength, and few examples where it increases bone strength, yet the trait can still have tremendous adaptive value for its host. Calcium homeostasis and skeletal lightness are two benefits of remodeling that confer no obvious advantage to the skeleton’s biomechanical integrity and are likely to reduce bone strength. Yet even when serving metabolic needs, features of the remodeling trait can minimize resulting increases in fracture risk. For example, when remodeling is activated to provide extracellular calcium or to eliminate underloaded bone, osteoblasts usually continue to refill the resulting resorption spaces with new bone matrix ( Fig. 31.3 ). Refilling under these two conditions might seem metabolically counterproductive, but without some degree of refilling, bone that is necessarily resorbed away to unlock calcium reserves or to decrease energy expenditure could lead to progressive and irreversible structural decay, with potentially dire consequences to mechanical function.
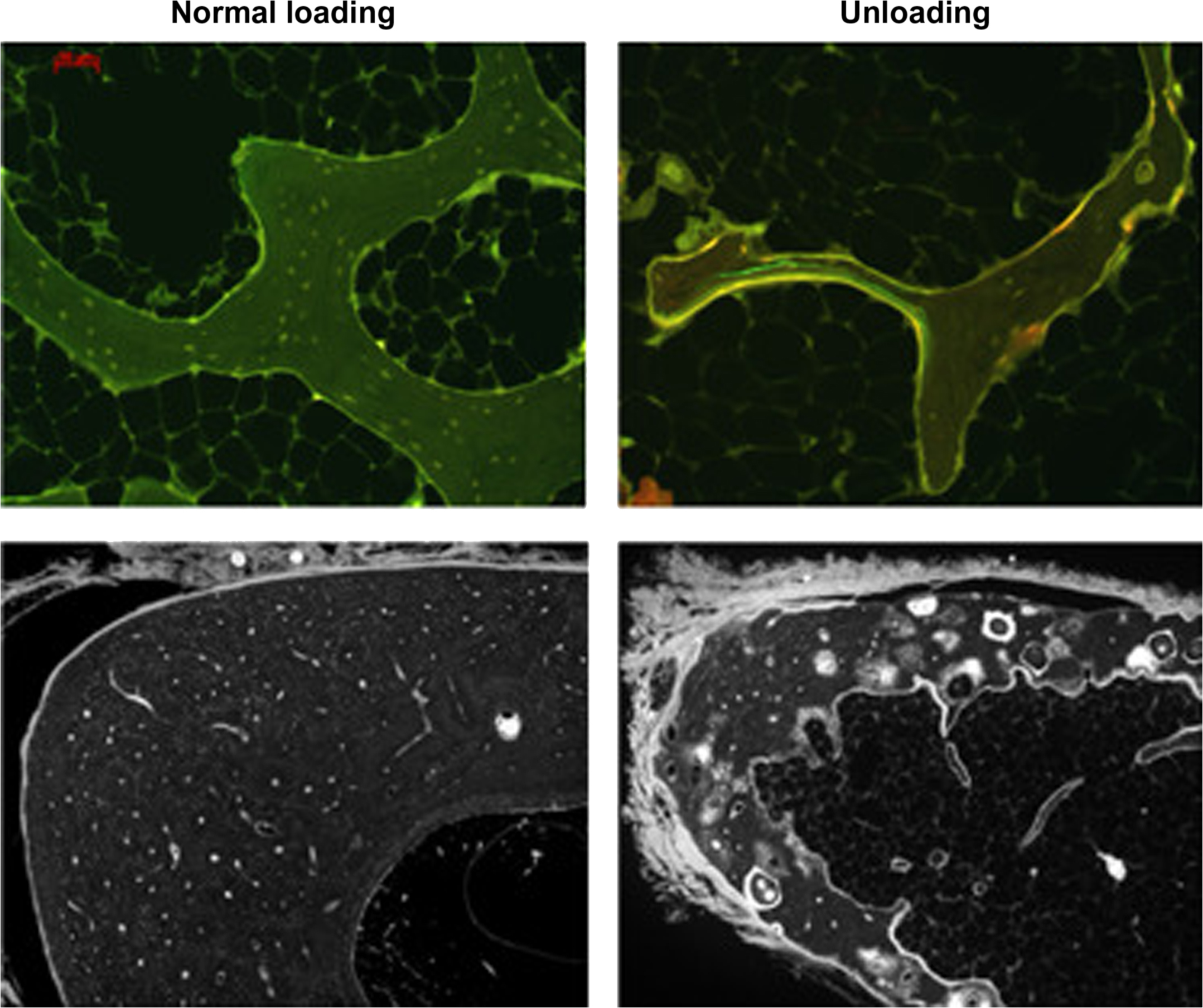
In the course of frequent conflicts between a host’s metabolic needs and bone structural strength, bone strength is usually sacrificed first. The evolutionary conservation of this trade-off suggests an overall benefit to the host’s survival and reproductive success, even if fracture risk might increase. (Perhaps unsurprising from an evolutionary perspective, there are few circumstances where the skeleton’s metabolic roles will provoke reductions in bone strength to the point that fracturing becomes inevitable.) Two examples highlighted later ( Sections 31.4 and 31.5 ) illustrate how remodeling activity can meaningfully reduce bone mass and strength in the service of metabolic imperatives. Section 31.9 then attempts to reconcile this biology with the commonly held view that continuous bone remodeling serves to maintain bone strength and reduce fracture risk. The “common view” is that remodeling prevents fractures by renewing bone matrix. An alternative view is that remodeling prevents fractures simply via the refilling of resorption spaces, with minimal regard to the mechanical competence of matrix that is removed. In this paradigm the main biomechanical benefit of remodeling relates to its ability to minimize strength losses , while the skeleton serves important metabolic imperatives.
31.4
The biological imperative of lightness
Relative to most extinct hominid species, we Homo sapiens have lithe, gracile skeletons. Neanderthals had much more robust skeletons that nonetheless fractured with remarkable frequency in adult males due to lifestyle, particularly crude hunting techniques . To regularly endure such punishment suggests an ability to heal a significant proportion of fractures, a major adaptive benefit in light of the relative scarcity of dietary resources and their substantial energy requirements. Body mass is an obvious determinant of caloric need and small reductions in skeletal mass have been suggested to confer meaningful energy benefits to a host . General weight-bearing and basal metabolic rate are two energetic considerations, and minimizing skeletal mass in the limbs makes them easier to accelerate and decelerate during locomotion. As stated by Currey, “It is clear that quite small savings in mass in the distal bones will be selected for strongly in animals whose power requirements for locomotion are important” .
Very high bone mass may be beneficial from the narrow perspective of fracture avoidance, but the overall costs of excessive bone mass are such that nature generally selects against this trait. BMU remodeling is an important trait ensuring that skeletal mass and strength are no greater than what is necessary to serve daily biomechanical functions, with an added safety factor to protect against some occasional traumas. Frost’s “mechanostat” theory proposes myriad ways that remodeling and modeling activities can adjust bone mass to match prevailing loading conditions . As described succinctly by Wang, “Bone modeling deposits bone mineral where it is needed and remodeling removes it from where it is not, to optimize strength while minimizing mass” . A discussion on the partnership of modeling and remodeling in skeletal optimization is found in Section 31.14 .
It is not remodeling per se but rather the osteoclast that specifically reduces the volume of underloaded bone, and increased bone resorption is among the more consistent findings during skeletal disuse, manifesting as a greater number of resorption spaces and/or increased resorption depth . Some animals, particularly those still growing, can also respond to disuse by decreasing bone formation on modeling-based surfaces, such as the long bone periosteum . This adaptation does not reduce bone mass in absolute terms, but it diminishes bone accrual and leads to a “less heavy” skeleton over time. Through the combined effects of greater resorption and decreased modeling-based bone formation, underloaded long bones can exhibit remarkable deficits in cortical including periosteal bone, leading to meaningful reductions in cross-sectional area and bending strength . Trabecular and endocortical surfaces of underloaded bone can also exhibit decreased bone formation, which might indicate a delay or reduction in the refilling of resorption spaces . This approach can lead to a lighter skeleton over time as new resorption spaces continue to be created but then slowly or incompletely refilled.
Some skeletal disuse studies also indicate seemingly paradoxical increases in bone formation parameters, a hallmark of increased bone remodeling . Increased remodeling rate will temporarily decrease bone mass via the remodeling transient ( Section 31.2 ), and if bone balance within BMUs is also negative due to disuse, the rate of bone loss will further increase, along with the risk of permanent structural deterioration in the form of composite osteons, cortical trabecularization, and trabecular perforations . Increased bone formation on remodeling surfaces of underloaded bones likely reflects attempts to refill a greater number of resorption cavities. Partial refilling of resorption spaces explains why increased bone formation can be evident even as the mass and strength of underloaded bones are in sharp decline ( Fig. 31.3 ) . This refilling might seem metabolically paradoxical if the goal is an overall reduction in bone mass, but some refilling might be important to minimize strength losses. Even partial refilling of deep, scalloped resorption cavities with a smooth covering of mineralizing osteoid could improve stress distribution in a manner that decreases the likelihood of crack initiation .
When remodeling is used to remove underloaded bone, the skeleton also remains well positioned for rapid structural recovery should normal loading resume. Many disuse settings are temporary, such as with illness or injury, and any resulting bone loss could leave a reloaded bone vulnerable to fracture. With BMU remodeling, resorption spaces that effectively lighten the skeleton will also serve as placeholders for future refilling, allowing for the rapid regain of bone mass and the preservation of original architecture and geometry. In monkeys subjected to disuse, an increased number of deep resorption spaces remained largely unfilled until reloading occurred, which provoked their rapid refilling in a manner that largely restored skeletal mass and original structure . These findings suggest a frequently overlooked benefit of continuous skeletal remodeling: during periods of disuse, a simple delay in the refilling of existing resorption spaces, combined with the ongoing creation of new resorption spaces, can rapidly and efficiently reduce skeletal mass and energy expenditure.
It seems unlikely that a light skeleton is the prime directive of BMU remodeling, or that this contribution explains its earliest evolutionary origins. This particular role does, however, indicate one-way remodeling provides metabolic benefits to a host even when the result is a reduction in bone strength and increased fracture risk. The next section describes the role of remodeling in calcium homeostasis, which represents a biological imperative so ancient and fundamental for life and reproduction that it might well account for the evolutionary adoption of continuous BMU remodeling.
31.5
The biological imperative of calcium homeostasis
Some key biological imperatives must be managed above other needs to ensure a host’s survival and reproduction. For example, despite the importance of skeletal muscle and internal organs, some animals routinely catabolize these tissues to serve the greater imperatives of energy security and hydration . Calcium homeostasis is another of the most ancient and critical biological imperatives, with blood calcium levels described as a “biological constant”: blood calcium concentration is similar and its normal range is very narrow in mammals, birds, reptiles, amphibians, and fish . Animals will undergo remarkable changes to maintain normocalcemia in the interests of preserving muscle contractility, blood coagulation, reproduction, and many other vital functions.
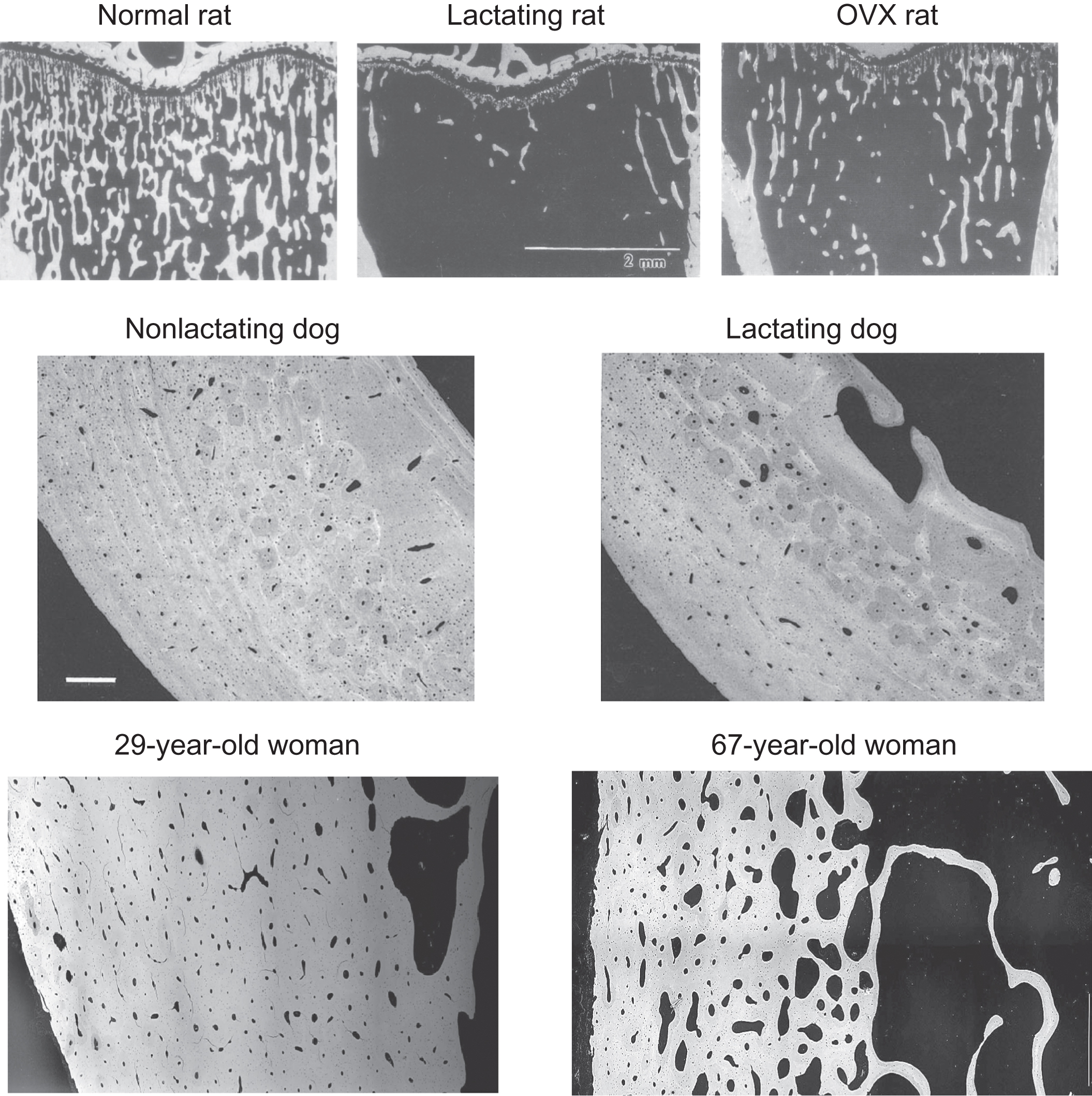
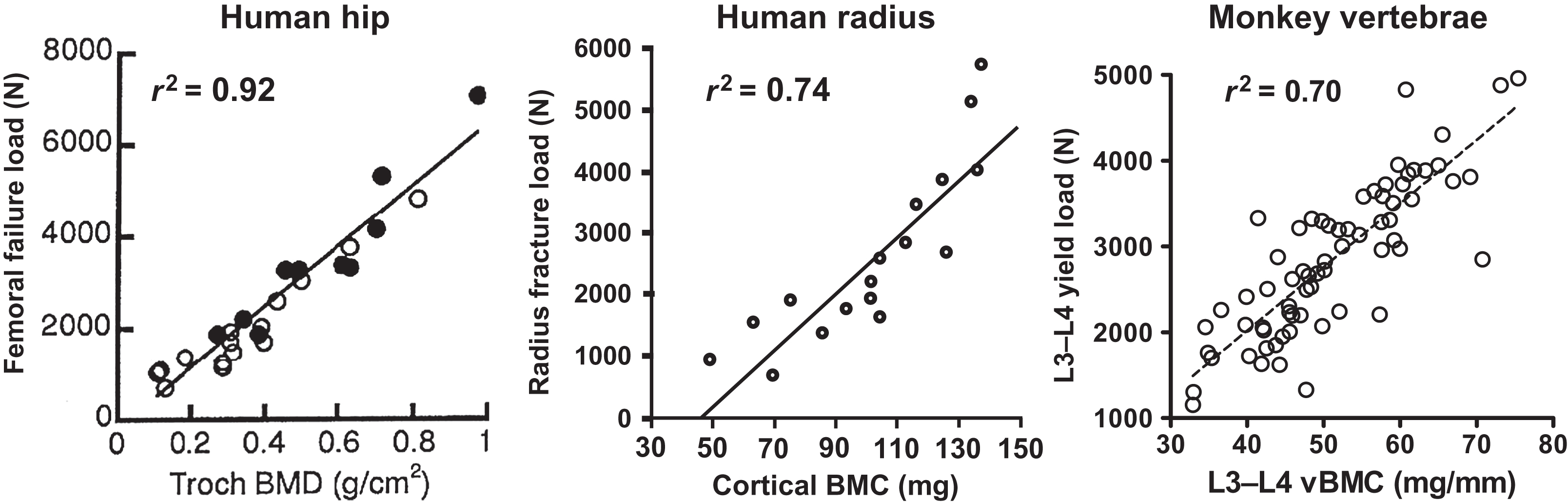
Virtually, all animals have calcium-storing organs or tissues as one strategy for ensuring the continuous availability of calcium. Fish store accessible calcium in their scales , some amphibians store calcium in endolymphatic sacs , and certain crustaceans store calcium in gastroliths . However, for the majority of terrestrial vertebrates and aquatic mammals, almost all internal calcium is stored in bone matrix. The universality of calcium storage is consistent with the likelihood that adequate dietary calcium is not always available in the natural world. Even short-term dietary calcium deprivation could compromise survival and reproduction were it not for accessible internal stores. The mobilization of skeletal calcium is probably the most reliable way to avoid or correct hypocalcemia in the natural world, even though it carries the drawback of reducing bone strength. By first principles, skeletal calcium mobilization inevitably decreases bone mineral content (BMC). With BMC being one of the best predictors of bone strength ( Fig. 31.4 ), the tapping of skeletal calcium reserves is very likely to decrease bone strength, and experimental data support this notion . Even minor increases in skeletal calcium mobilization should reduce bone strength, though to a degree that may only be measurable through the harsh verdict of natural selection, in the form of a subtle excess in traumatic fractures that reduce lifespan or reproductive potential. Despite the likelihood that any reduction in bone strength increases the risk of fractures in the natural world, increased fracture risk appears acceptable when calcium homeostasis is at stake. Recognizing this trade-off, Herbert Fleisch wrote, “Since 99% of the body calcium is located in the skeleton, this will act as a reserve of the ion. In the case of calcium shortage, the homeostatic mechanisms will work to the detriment of bone in order to maintain calcium, which seems to have absolute priority” . Remarkable findings from genetically modified mice demonstrated their ability to avoid hypocalcemia through severe sacrifices of bone mass and strength, supporting the investigators’ conclusion that “the cellular processes regulated by serum calcium will be guaranteed at the expense of skeletal integrity, even when these adaptations result in bone fractures” .
The skeleton can contribute to calcium homeostasis in ways other than via remodeling, but reduced bone strength remains a common result. For example, calcium deprivation in young growing animals can impair osteoid mineralization, leading to profoundly reduced bone strength . Immature animals can also reduce the growth rate of their skeletons to conserve extracellular calcium . Smaller stature and a weaker, osteomalacic skeleton are apparently worthwhile sacrifices when they foster maintenance of blood calcium. In skeletally mature humans, skeletal contributions to calcium homeostasis are primarily and perhaps exclusively mediated by BMU remodeling. Decades of uncertainty regarding this assertion were sustained in part by investigators’ early reliance on agents that inhibit osteoclasts less strongly and less rapidly than those available today. Early-generation bisphosphonates can take days or weeks to maximize their osteoclast inhibitory effects, which provides the parathyroid hormone (PTH) and vitamin D axis ample time to avoid hypocalcemia by increasing fractional calcium absorption in the gut and calcium reabsorption in the kidney . Further obscuring the important role of osteoclasts in calcium homeostasis is the unnaturally high calcium content of standard laboratory animal diets, which are typically provided ad libitum . Equipped with a limitless supply of dietary calcium, bone remodeling and skeletal reserves are probably unimportant for maintaining normocalcemia.
The extent to which BMU remodeling mobilizes skeletal calcium came into sharper focus when recombinant OPG and RANKL became available as research tools. These reagents rapidly and selectively regulate osteoclast differentiation, activity, and survival ( Fig. 31.1 ) . Early animal studies show that OPG caused greater and more rapid reductions in serum calcium compared with the effects of parenterally administered bisphosphonates . The relationship between osteoclast activity and blood calcium can be so consistent that early pharmacology studies of RANKL inhibitors often relied on decreased serum calcium as a surrogate for osteoclast inhibition . The initial in vivo screen used to identify RANKL antibodies for clinical development relied on their ability to prevent RANKL-induced (i.e., osteoclast-mediated) hypercalcemia in normal mice . When the selected candidate (denosumab) was tested clinically, it was ultimately shown to reduce serum calcium to a greater extent than oral or intravenous bisphosphonates .
The availability of RANKL inhibitors provided new ways to address whether osteoclasts can rapidly respond to hypocalcemia, or only over longer timeframes, as some have suggested . PTH secretion is the immediate response to hypocalcemia, and PTH injections in birds increased plasma calcium and osteoclast differentiation features within 20 minutes . In mice, injection of PTH or RANKL increased serum calcium within 60 minutes . RANKL increased osteoclast numbers in mice within 6 hours of injection , and the hypercalcemic effects of RANKL can be sustained from 3 days to 6 weeks . Conversely, OPG and other RANKL inhibitors reduced serum calcium after 2 hours , 3 hours , 4–6 hours , 16–30 hours , 5 days , 14 days , 1 month , 3 months , and 16 months . Based on these and other studies, it is difficult to identify times and conditions where osteoclasts are not capable of contributing to calcium homeostasis, though minute-to-minute calcium regulation may be of a different domain. Several studies suggest that osteocytes, those terminally differentiated osteoblasts that form a network throughout bone matrix, can resorb matrix within their lacunae . It is unclear whether this phenomenon leads to meaningful transfer of calcium into blood, and this question is somewhat confounded because most conditions associated with lacunar resorption are also associated with osteoclastic resorption. Regardless, there is evolutionary appeal to the notion that a nonosteoclastic bone cell might also be capable of mobilizing skeletal calcium reserves when needed.
In normal healthy animals and humans, even rapid and profound osteoclast inhibition rarely causes symptomatic hypocalcemia, which says more about the importance of maintaining calcium homeostasis than it does about remodeling’s role in serving that imperative. Calcium security is assured by the joint contributions of BMU remodeling, intestinal calcium absorption, and renal calcium reabsorption. This arrangement can be conceptualized as a stool with three legs, the minimum number needed for stability. Without BMU remodeling, calcium security would rely on the vagaries of dietary calcium availability and the kidney’s ability to minimize calcium losses. Diet may be the preferred means of maintaining calcium, but this approach is hardly failsafe. One safeguard that may enhance dietary calcium security is the presence of functional calcium-sensing taste receptors on the tongue of humans and mice . Yet this trait, and some other important homeostatic processes, remains reliant on the availability of some dietary calcium. Fractional calcium absorption in the gut increases with hypocalcemia or decreased dietary calcium intake , but this adaptation has diminishing value when dietary calcium is scarce or unavailable. Meanwhile, hypocalcemia is associated with appetite suppression, fatigue, confusion, cramping of hands and feet, bone pain, bronchospasm, tetany, and seizures , all of which are likely to cruelly diminish a host’s ability to scavenge for calcium-containing food (and avoid predation). Renal calcium reabsorption, which is stimulated by the PTH response to hypocalcemia, is another important homeostatic mechanism that decreases calcium losses in the urine, but this response has limited ability to cause absolute increases in blood calcium, and it does not mitigate obligatory calcium losses in feces and sweat .
Thus while dietary calcium and renal calcium reabsorption are very important for calcium homeostasis, their combined impacts cannot assure perpetual normocalcemia. Even temporary failure of either system could lead to a host’s demise, were it not for bone remodeling. When hypocalcemia triggers the remodeling system, net transfer of calcium into blood is virtually assured. This transfer may or may not occur within minutes, but it certainly occurs in under an hour and can persist for weeks or months. The skeleton is a virtually failsafe source of calcium, which may explain why the maternal skeleton of humans and countless other species is relied upon to provide much of the calcium used for the most important evolutionary imperative of all, reproduction ( Section 31.10 ). Consistent with these concepts, potent antiresorptives can cause significant and symptomatic hypocalcemia in states of high calcium demand, such as with growth, osteomalacia, and lactation . Strong remodeling inhibition can also lead to symptomatic hypocalcemia in patients with severely reduced renal function who are not adequately supplemented with dietary calcium .
If osteoclasts are not vitally important to calcium homeostasis unless other regulatory systems fail or calcium demand spikes, why does bone remodel continuously throughout life? An alternative “on-demand” system, wherein remodeling is activated only when needed, might be more sparing of skeletal mass and more energy efficient. However, when hypocalcemia strikes, an on-demand arrangement may be too sluggish to confer meaningful advantages to short-term survival. It takes several days for osteoclast precursors to mobilize, differentiate, fuse, attach to bone, and resorb mineralized bone matrix to mobilize calcium. In contrast, our continuous remodeling arrangement provides a large and constant supply of mature bone-resorbing osteoclasts that can readily respond to hypocalcemia simply by increasing their activity or survival. These responses may explain the ability of RANKL to increase blood calcium within an hour of its injection . The millions of BMUs available in the continuously remodeling skeleton also provide a “calcium sink” in the form of osteoid and immature mineralizing bone matrix, which would not be perpetually available with an on-demand remodeling arrangement. The value of this calcium sink is highlighted by the challenges of some renal disease patients, who have minimal osteoid, in avoiding hypercalcemia with increased dietary calcium intake . Conversely, some patients with primary hyperparathyroidism have excessive osteoid that can increase the risk of hypocalcemia after parathyroidectomy. This “hungry bone syndrome” is thought to reflect the ongoing mineralization of extensive osteoid that had previously been serviced by excessive PTH-stimulated bone resorption . Just as financial markets require a substantial flux of capital to operate efficiently, the efficiency of calcium homeostasis is largely predicated on the constant and regulatable flux of calcium provided by continuous BMU remodeling.
31.6
Calcium, remodeling, and skeletal structure
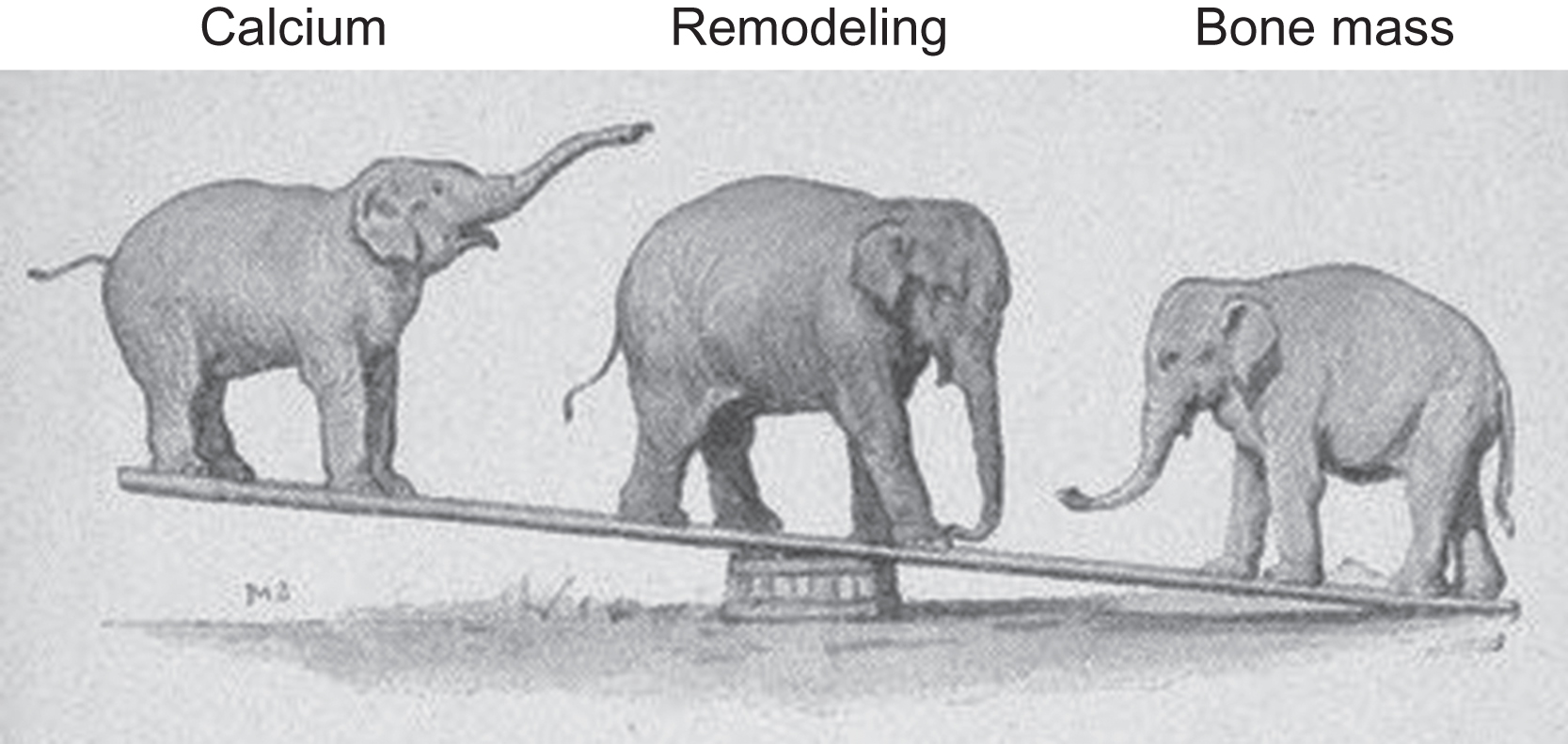
Starting with a crude evolutionary framework, when calcium demands and bone structural needs collide, calcium demands usually win or the host’s genes might lose. At the nexus of this relationship is bone remodeling, which increases with calcium demand at the expense of bone structure. When calcium demand declines, BMU remodeling typically declines as well, to the benefit of skeletal structure. These relationships are depicted in Fig. 31.5 . The elephant on the left represents calcium demand, or extracellular calcium concentrations in blood, urine, and/or breast milk. The calcium elephant will rarely represent calcium demand and extracellular calcium, because when calcium demand increases, extracellular calcium in various compartments frequently declines. However, either calcium demand or extracellular calcium concentrations should change in accordance with the cartoon. The elephant at the lever’s fulcrum represents the remodeling apparatus, and the elephant at the other end represents bone mass (and, frequently, bone strength). Befitting its status as a major biological imperative, the calcium elephant appears to aggressively instruct the remodeling elephant to move forward or backward at its bidding, causing predictable changes in calcium and bone mass. Increased calcium demand increases remodeling, and bone mass decreases as remodeling mobilizes skeletal calcium into blood. Antiresorptive agents decrease remodeling activation, and as preexisting remodeling space refills and mineralizes, bone mass increases while extracellular calcium levels (typically in blood and/or urine) decline. As another example, the direct activation of remodeling via the proresorptive cytokine RANKL increases extracellular calcium at the expense of bone mass. The following sections describe these and other examples of inverse relationships between calcium and bone mass. Remodeling mediates such conflicts in a manner that almost always favors calcium, but does so in ways that minimize the biomechanical impact of reduced bone mass. Particular attention will be paid to the bone-forming phase of BMU remodeling, to highlight the importance of refilling resorption spaces even in states where the apparent goal of remodeling is to furnish extracellular calcium.
31.7
Effects of pharmacologically modified remodeling on calcium and bone mass
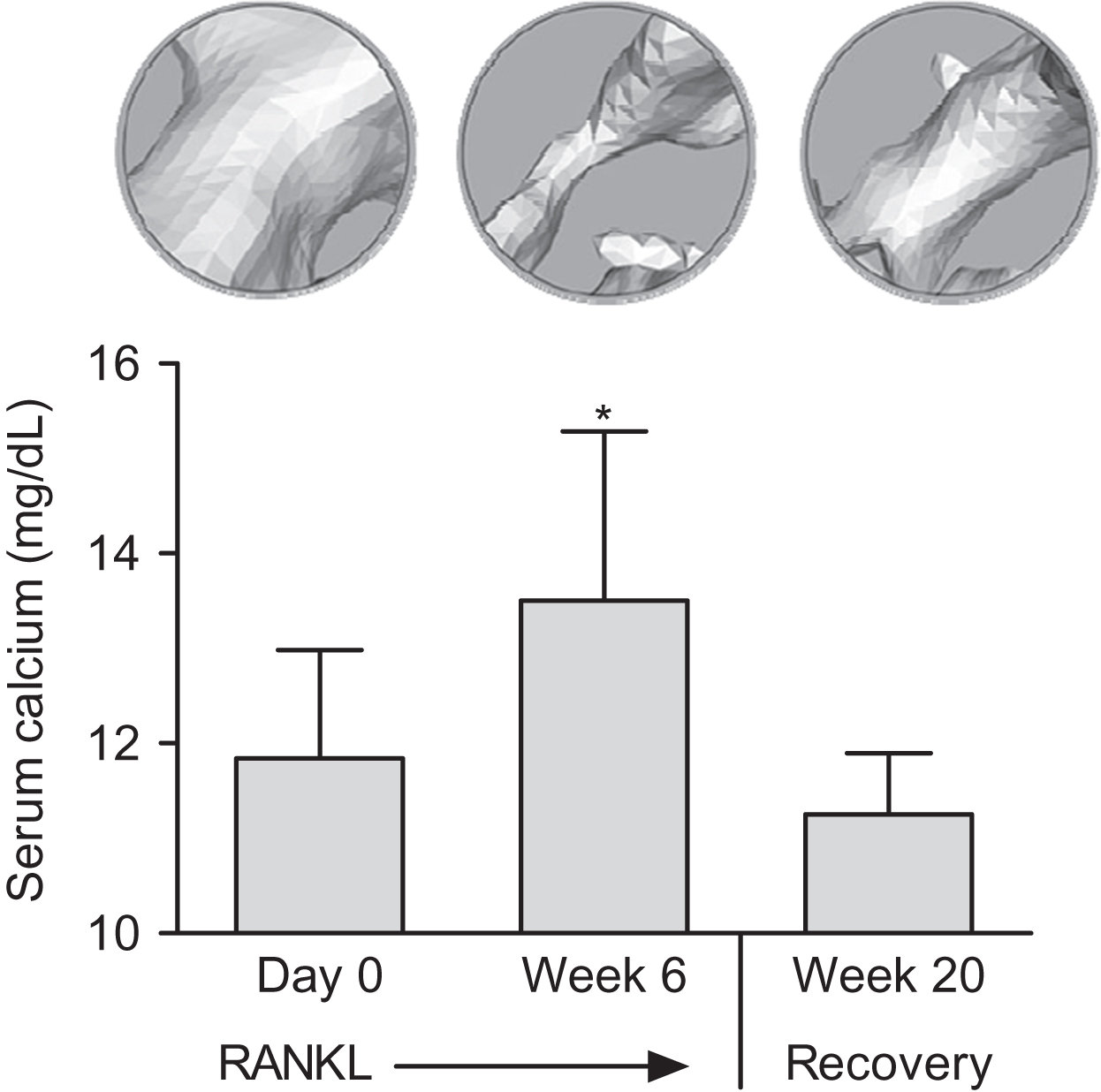
Administration of proresorptive or antiresorptive agents can affect remodeling in ways that predictably alter extracellular calcium and bone mass in accordance with Fig. 31.5 . RANKL and RANKL inhibitors will serve as examples here because they operate through physiological pathways and exert potent and opposing effects on remodeling. A physiological role for RANKL in calcium homeostasis is supported in many studies, including evidence for rapid upregulation of skeletal RANKL expression during dietary calcium deprivation . Direct injections of recombinant RANKL may mimic an increase in calcium demand, and RANKL injections rapidly activate osteoclasts to initiate BMU remodeling, which increases serum calcium while reducing bone mass . When RANKL injections cease, serum calcium returns to normal and some of the lost bone mass and volume are restored ( Fig. 31.6 ) . The latter result highlights the key “placeholder” benefit of the BMU remodeling trait, in that much of the bone lost with skeletal calcium mobilization can eventually be replaced in the same location, thereby preserving much of the original bone geometry and architecture. RANKL can also cause irreversible loss of bone mass and strength due to trabecular perforations, loss of entire trabecular elements, and coalescence of endo- and intracortical porosities ( Fig. 31.7 ) . Some of this permanent bone loss might also relate to stimulation of resorption drifts that are not destined for refilling .
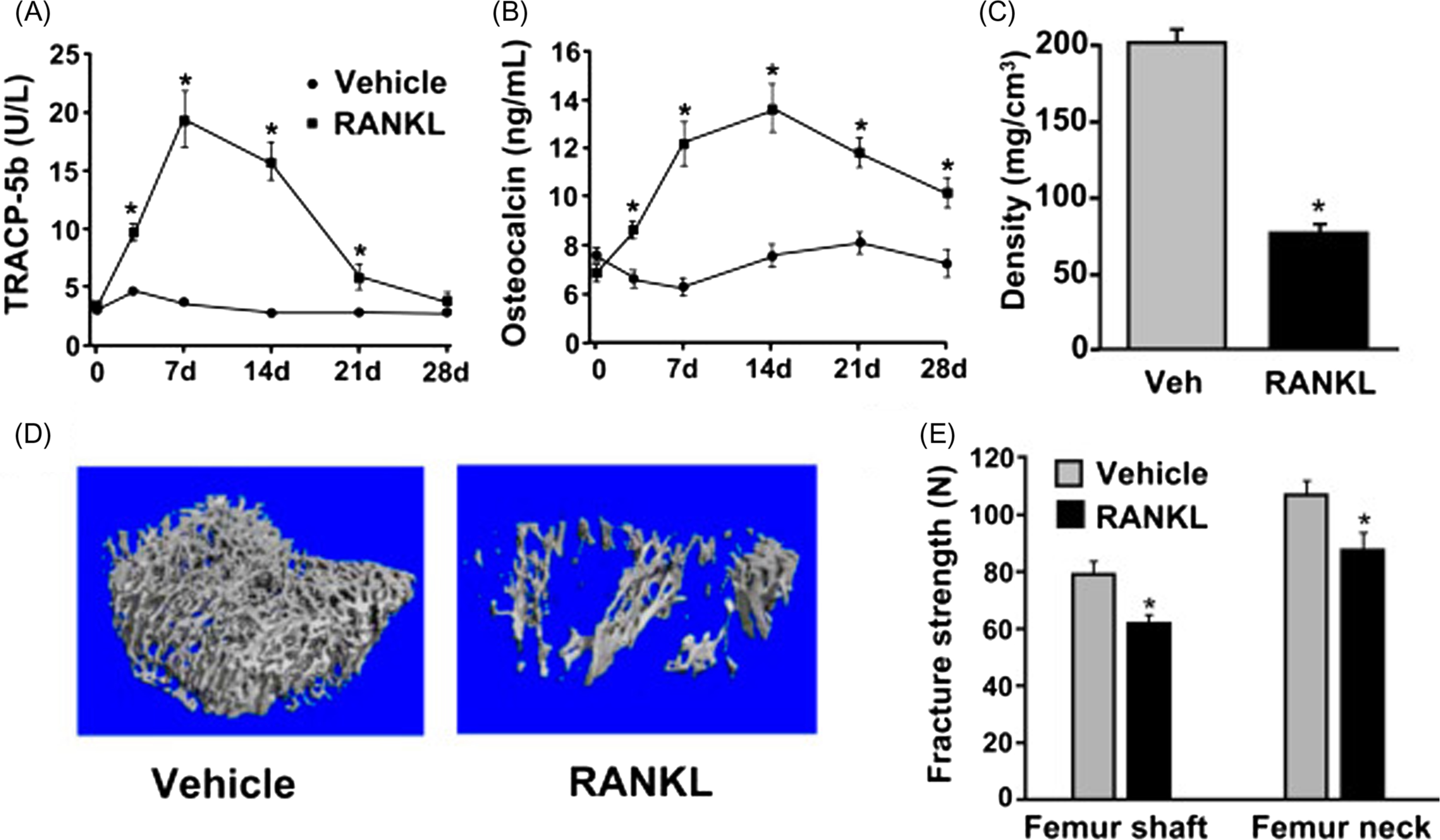
RANKL does not act directly on osteoblasts but will nonetheless induce increases in bone formation, whether by its direct injection or by ablation of OPG, its natural inhibitor. This increase in bone formation, which indicates the refilling of a greater number of resorption spaces, is evident even as bone mass and strength are in decline ( Fig. 31.7 ) . Without some refilling of resorption spaces, bone strength losses induced by RANKL would likely be even more severe. Remodeling-associated increases in bone formation are common even in settings where the reduction of bone mass (e.g., during disuse) or the extraction of bone mineral (e.g., during lactation) appears to be the very goals of BMU remodeling. However, even when refilling does not keep pace with resorption, some refilling might be biomechanically advantageous in terms of blunting the stress-raising impact of deep scalloped resorption lacunae and intracortical porosities . Mechanical failure of human trabecular bone has been suggested to initiate preferentially near resorption cavities , and by eventually refilling these cavities, BMU remodeling might minimize strength losses while other priorities are calling for reductions in bone mass.
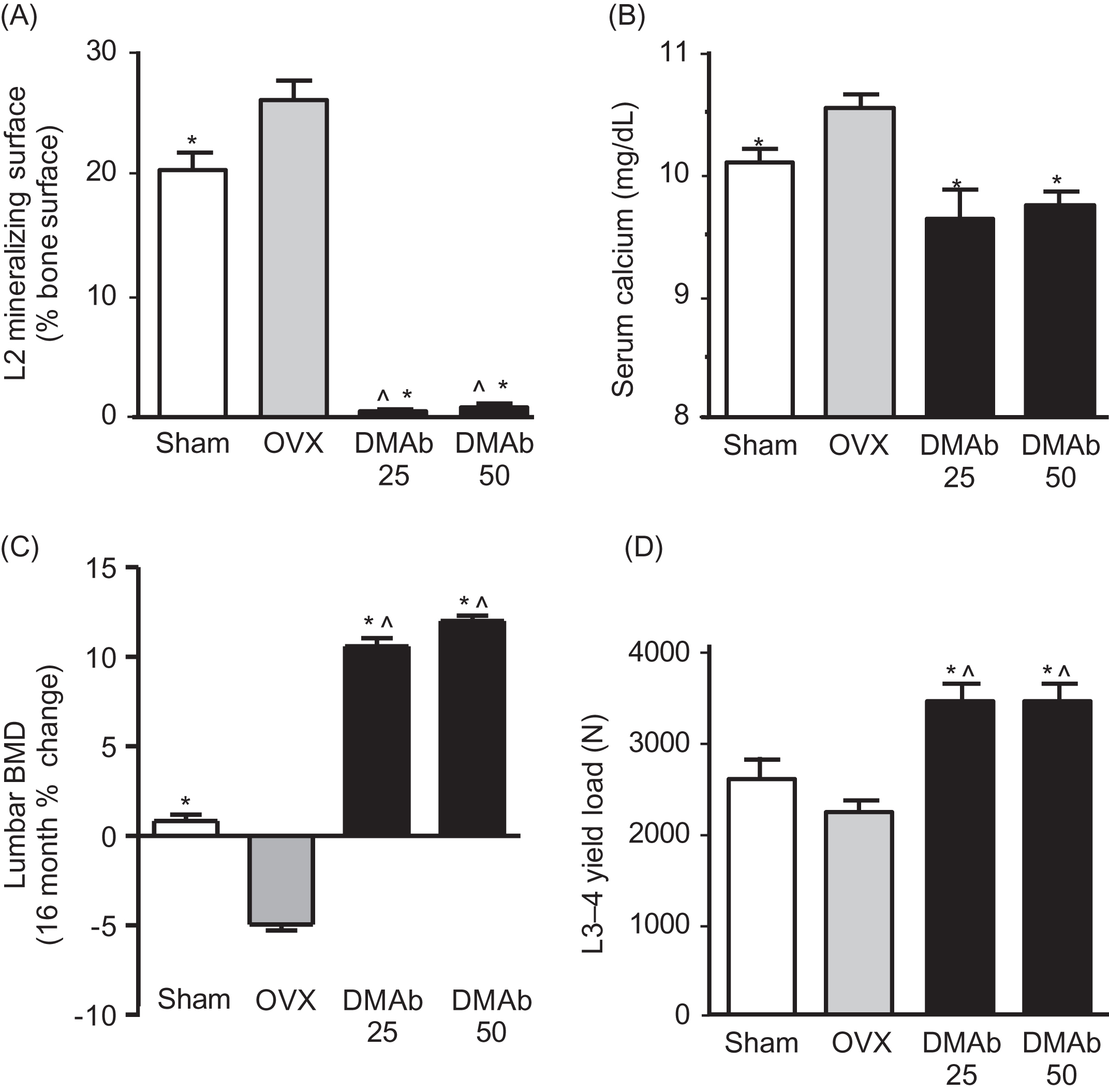
RANKL inhibitors such as OPG and denosumab rapidly decrease osteoclast activity, which often leads to reduced serum and urine calcium. This is followed by decreased bone formation, which indicates the successful completion of preexisting remodeling cycles that will leave fewer resorption spaces in need of refilling. The end result is increased bone volume, mass, and strength . Fig. 31.8 illustrates these changes in a study of cynomolgus monkeys treated for 16 months with the RANKL inhibitor denosumab. When remodeling was increased by ovariectomy or decreased by denosumab, remodeling and serum calcium changed in one direction and bone mass the other, in accordance with Fig. 31.5 .
31.8
Effects of dietary calcium on bone mass and strength
In postmenopausal women, many of whom have inadequate calcium intake secondary to decreasing fractional calcium absorption , calcium supplements can decrease BMU remodeling and increase bone mass , in accordance with Fig. 31.5 . Similar responses occurred after calcium supplementation in Gambian children accustomed to a low-calcium diet . Calcium supplements decreased bone formation indices in both studies, an indirect suggestion that fewer resorption spaces were being created and refilled after the increase in dietary calcium intake. Greater dietary calcium intake can also enhance bone mineral density (BMD) gains in healthy young women , suggesting their habitual bone mass had been limited by real or perceived calcium deficiency. Dietary calcium deficiency can activate BMU remodeling, leading to bone loss that helps to maintain blood calcium. Rats on a low-calcium diet exhibit increases in serum PTH, skeletal RANKL messenger ribonucleic acid (mRNA) expression, fractional calcium absorption, renal calcium reabsorption, and BMU remodeling, along with decreased cortical and trabecular bone mass . These responses can usually prevent hypocalcemia, but further increases in calcium demand such as during lactation can upset this delicate balance, and hypocalcemia can result .
Just as bone formation can decrease after calcium supplementation, bone formation parameters can increase with calcium deprivation and with impaired intestinal calcium absorption . Increased bone formation in these settings likely reflects the refilling of additional resorption spaces created to furnish extracellular calcium. This refilling, even if complete, should lessen the adverse biomechanical impact of the extra resorption space by smoothing over jagged resorption surfaces. Another adaptation available to a calcium-deprived host is to delay the mineralization of osteoid , which confers two benefits: relatively more calcium remains available in the blood, while the unmineralized osteoid serves as a placeholder for future mineralization and restoration of original architecture. This approach might have biomechanical advantages over the theoretical alternative of creating fewer resorption spaces that never refill a possible scenario for a host without postcranial BMUs.
Two genetically modified mouse strains provide a glimpse into a hypothetical world where BMU remodeling did not evolve as a response to hypocalcemia. PTH knockout mice cannot rely on the normal physiological trigger (PTH release) by which the skeleton, gastrointestinal tract, and kidney maintain blood calcium, and these mice exhibit hypocalcemia despite a standard calcium-rich laboratory diet . Unable to remodel bone in response to hypocalcemia, PTH knockout mice exhibit significantly fewer osteoclasts, less bone formation, and much greater cortical and trabecular bone mass relative to wild-type controls. Similarly, mice lacking a key genetic control region that regulates RANKL mRNA expression exhibit diminished remodeling responses to PTH and 1,25(OH) 2 D 3 , and these mice also exhibit less constitutive remodeling and greater bone mass and bone strength than wild-type controls . Greater bone mass and strength in these mutant mice would probably reduce their risk of fractures in the natural world, but the energy costs of carrying the extra mass would be a potential liability, as would the chronic risk of hypocalcemia. Nature apparently selected skeletons that are able to remodel when calcium is needed, at the expense of bone mass and strength.
In addition to calcium intake, the ability to absorb intestinal calcium can also regulate bone remodeling, bone strength, and fracture risk. Elderly humans often have reduced serum vitamin D levels and lower fractional calcium absorption, which can activate remodeling via hypocalcemia and PTH responses that have been linked to age-related bone loss and increased fracture risk . Bone loss in the elderly can be exacerbated by a negative bone balance , with reflects a state of incomplete refilling that will also lead to a greater net mobilization of skeletal calcium with each remodeling cycle. Based on the priority of calcium homeostasis over fracture avoidance, a negative bone balance could potentially have some adaptive value in the elderly: incomplete refilling could compensate for their reduced fractional calcium absorption, helping to maintain blood calcium in a manner that also compromises skeletal integrity. Animal studies show that the skeleton can suffer remarkable deterioration when calcium is poorly absorbed. For example, selective ablation of the vitamin D receptor in the intestine of mice greatly diminished dietary calcium absorption, but skeletal adaptations allowed them to remain normocalcemic. These adaptations included increased bone remodeling, decreased osteoid mineralization, and reductions in bone mass so severe that spontaneous fragility fractures were observed . The phenotypes of two inbred mouse strains with substantial genetic divergence in vitamin D metabolism also seem to demonstrate this inverse relationship between calcium absorption and bone strength. C3H/HeJ mice exhibit higher serum 1,25(OH) 2 D 3 , greater fractional calcium absorption, and higher serum calcium than the C57Bl/6J strain. As predicted in Figs. 31.5 , C3H/HeJ mice also have more robust skeletons than C57Bl/6 mice, including greater BMD, bone calcium content, cortical thickness, and bone strength .
Just as calcium absorption can regulate remodeling, remodeling can regulate calcium absorption. Remodeling inhibition in alendronate-treated postmenopausal women was accompanied by increases in serum PTH and 1,25(OH) 2 D 3 , which presumably explained an observed doubling of their fractional calcium absorption . Similarly, strong remodeling inhibition in denosumab-treated cynomolgus monkeys triggered increases in serum PTH and 1,25(OH) 2 D 3 . In accordance with Fig. 31.5 , the denosumab-treated animals exhibited decreased blood calcium and increased bone mass and strength, and it is likely that blood calcium would have declined more substantially were it not for the serum PTH and vitamin D responses triggered by denosumab.
31.9
An evolutionary theory on the origin of continuous bone remodeling
Gould’s evolutionary framework suggests that even the most highly conserved traits can serve important purposes in less-than-perfect ways. BMU remodeling might not be the optimal way to provide calcium from the narrow perspective of bone biomechanics, but it proved good enough to solve one of evolution’s greatest challenges, as described later. Suboptimal traits abound in nature because evolution is an incremental process, building upon earlier traits that can leave a host with imperfect but adequate solutions that undergo positive selection. Meanwhile, parallel traits often emerge to compensate for the shortcomings of original traits. BMU remodeling and the quasiparallel bone modeling system ( Section 31.14 ) represent possible examples where an evolutionary challenge (calcium homeostasis) was met by coopting an existing trait (jawbone remodeling) and applying it throughout the skeleton, which imperfectly met the original challenge while creating a new problem (bone loss). In turn, another ancient trait (bone modeling) may have been coopted in compensatory fashion to better maintain skeletal integrity. This arrangement would allow the skeleton to serve as a calcium reservoir with minimal increases in skeletal fragility over our natural lifespan. Nature might be somewhat indifferent whether this arrangement remains effective in advanced age or beyond the menopause, which could explain increased fracture risk in those populations.
Metabolic reliance on skeletal calcium is a relatively late, derived evolutionary trait . “Late” could mean about 400,000,000 years ago, when vertebrates began their invasion of land to become tetrapods (i.e., four-limbed descendants of lobe-finned vertebrate fish, including all amphibians, reptiles, dinosaurs, birds, and mammals) . “Derived” is a contradistinction from original ancestral traits, and the original traits for managing calcium homeostasis in early aquatic vertebrates including fish did not appear to leverage skeletal calcium. This supposition is partly based on the biology of modern teleost (ray-finned) fishes, which share a recent common ancestor with the lobe-finned fish from which all tetrapods (including aquatic mammals) evolved. The great majority of modern fish species are ray-finned , but a few lobe-finned varieties persist, such as coelacanths and lungfish. Little is known about calcium homeostasis or skeletal biology in lobe-finned species, which is unfortunate because they might have interesting intermediate features between ray-finned fish and the amphibian class to which they gave rise. Based on limited data from frogs, amphibians seem to rely on skeletal reserves as mammals do when calcium is needed . In contrast, ray-finned fishes (henceforth called fish) can secure calcium in ways their terrestrial descendants cannot, such that the calcium stored within their mineralized skeletons is rarely if ever needed .
Fish do possess bone-resorbing osteoclasts, which are needed for growth-related modeling-based bone resorption and for tooth eruption, but most extracellular calcium in fish is provided by diet and by direct absorption of calcium ions from the water via their gills . Fish produce several PTH-like peptides but they lack parathyroid glands, which Melvin Moss considered logical because they should not need a coordinating center for calcium homeostasis when they swim in a near-infinite supply of readily absorbable calcium ions . Freshwater 1
1 Phosphate concentration is low in marine and freshwater, and diet is its main source.
has much less calcium than marine water , and freshwater can sometimes create calcium supply challenges that are managed via numerous remodeling-independent adaptations. For example, some freshwater species meet calcium demands by resorbing their calcium-rich scales , which may be mediated by PTH-induced RANKL expression . The extreme laboratory condition of acalcemic water combined with physical scale debridement was capable of inducing some endoskeletal bone resorption in fish, suggesting a latent capacity for accessing skeletal calcium reserves as an adaptation of last resort .These laboratory conditions aside, there is minimal evidence that fish in their natural habitats rely on their postcranial endoskeleton for calcium. “Postcranial” is a potentially important qualifier, because continuous remodeling activity is evident in the tooth-bearing bones of fish, and there is evidence that drastic increases in calcium demand in laboratory settings can increase this activity . However, the main physiological role of continuous remodeling in the fish jaw is not calcium homeostasis but rather to service repetitive tooth eruption and to maintain the attachment of teeth as they are constantly repositioned to accommodate perpetual jawbone growth. This jawbone remodeling activity exhibits numerous histological features of classical BMU remodeling, including the resorption of discrete packets of mineralized bone matrix by osteoclasts, followed by the deposition of a cement line and osteoblast-mediated refilling of resorption spaces with mineralizing osteoid . The temporospatial coordination of these BMU activities allows for incremental tooth migration without loss of attachment to the jawbone via ankylosis. A closely related trait is apparent in the jaws of many modern terrestrial vertebrates, including humans, with a key variation being the incorporation of Sharpey’s fibers into the cement line, which reconnects a periodontal ligament to alveolar bone so teeth remain attached to the perpetually dynamic tooth socket . It is conceivable that the jawbone remodeling traits expressed in ancient fish provided the evolutionary template for the BMU remodeling trait that is continuously applied throughout the skeletons of modern vertebrates. Evidence below suggests that challenges to calcium homeostasis provided the catalyst for this adoption, starting with the earliest tetrapods.
As mentioned previously, fish appear capable of increasing jawbone remodeling during extreme calcium deprivation , but there seem to be few natural scenarios where this latent capacity would be activated in the postcranial fish skeleton. Osteoclasts populating the postcranial fish skeleton are primarily focused on modeling and sculpting their perpetually growing bones, which is accomplished by small mononuclear osteoclasts that dig very shallow pits, a process called “smooth” resorption . Mammals also resorb some bone with these primitive mononuclear osteoclasts , but the major mammalian mode of surface resorption (trabecular, endocortical) involves large multinucleated osteoclasts that create deep scalloped Howship’s lacunae ( Fig. 31.2 ). Fish can create Howship’s lacunae in tooth-bearing bones under laboratory conditions of extreme calcium deprivation , and Howship’s lacunae have been observed in jawbones of freshwater species and in marine species that are transiently exposed to the low calcium of freshwater during spawning runs . The selective creation of Howship’s lacunae under these conditions suggests that this mode of resorption is preferable for calcium mobilization, whereas smooth resorption might be more suitable for the slower and more deliberate modeling (shaping) of growing bones. But whether smooth and shallow or deep and scalloped, there is minimal evidence that resorption spaces in the postcranial skeleton of fish are ever refilled, which indicates the general absence of remodeling.
Absent the remodeling trait, the fish endoskeleton can still contribute to calcium homeostasis. Most fish grow continuously throughout life, which provides a continuous modeling apparatus that can be exploited in times of extreme calcium deprivation. For example, fish maintained in acalcemic water on an acalcemic diet can maintain blood calcium by increasing osteoclastic resorption on bone surfaces that undergo perpetual resorption, while decreasing bone formation on surfaces undergoing perpetual apposition . Calcium repletion reverses these changes and also restores skeletal growth rate, which had been reduced by half during calcium deprivation. There was, however, no histological evidence indicating that the smooth, shallow resorption spaces created to maintain blood calcium were also being refilled. If such contrived environmental conditions persisted, a continued reliance on these modeling-based adaptations may further impair growth and may also progressively distort bone geometry. This inference highlights a key difference between modeling and remodeling: the former always alters while the latter tends to preserve bone shape and architecture .
It is hypothesized that fish do not refill resorption spaces for lack of need or consequence. The ability to directly absorb aqueous calcium through gills and to resorb calcium-rich scales would generally spare the endoskeleton from participation in calcium homeostasis. Furthermore, their continuous modeling apparatus may also be leveraged as a last resort to maintain extracellular calcium. Armed with these adaptations, deep scalloped resorption lacunae are rarely if ever created in the postcranial skeleton of fish, and if episodic spikes in calcium demand ever required their creation, the consequences of leaving them unfilled would be minimal because their buoyant environment protects them from fracture. The refilling of resorption spaces, which essentially defines BMU remodeling, would seem to be an indifferent trait for fish, unworthy of selection or conservation. Fish osteoblasts appear to ignore the occasional resorption space created in their postcranial skeleton, whereas terrestrial vertebrates that habitually create such resorption spaces also consistently refill them. 2
2 Modeling-based bone resorption, that is, resorption drifts, being a notable exception.
Everything changed when fish evolved into land-dwelling tetrapods. The first tetrapods that invaded land some 400,000,000 years ago were fish-like creatures with amphibian features, including primitive lungs, rudimentary limbs, and a lack of scales . These pioneers would face major challenges to calcium homeostasis as they gradually abandoned a limitless supply of directly absorbable calcium ions in favor of an unfamiliar land-based diet . A day or two of unsuccessful foraging for calcium on dry land could lead to hypocalcemia in these primitive tetrapod pioneers, some of which may have met this challenge by increasing modeling-based resorption and reducing modeling-based formation . However, continued reliance on this trait could lead to reduced growth or progressive distortion of skeletal geometry, the long-term consequences of which would be all the more severe for a terrestrial skeleton facing increased gravitational loads. Through random genetic mutations, tetrapods eventually applied the jawbone remodeling trait to their postcranial skeleton, thereby providing a new, efficient, and biomechanically favorable way to harvest calcium from bone matrix. This “drill-and-fill” approach, previously restricted to the tasks of tooth eruption and tooth movement, would provide a constant flux of extracellular calcium while also minimizing reductions in skeletal strength. This “tetrapod-calcium theory” remains consistent with the contemporary notion that bone remodeling is vital for the maintenance of bone strength. But in contrast to the current “matrix renewal” paradigm, remodeling’s contribution to bone strength under this alternative paradigm relates not to the (unfit) biomechanical competence of bone that is removed , but to the simple fact that the resorbed bone (fit or unfit) is eventually replaced.
Matrix renewal could be teleologically invoked as an evolutionary driver of continuous bone remodeling in terrestrial tetrapods based on the added gravity their skeletons experienced on land. For example, higher gravitation loads that increased skeletal strain may have caused more wear-and-tear-related damage than their aquatic forbearers experienced, and remodeling-based matrix renewal could mitigate damage accumulation and thereby reduce the risk of structural failure. In considering this premise, it should first be noted that increased skeletal strain experienced by early terrestrial vertebrates was probably managed over time by natural selection of more robust and stiffer skeletons, adaptations that would be achieved via modeling -based adaptations that regulate bone size, shape, and overall geometry. Despite such adaptations, terrestrial life can still lead to skeletal damage, and bone remodeling is uniquely capable of removing microscopically damaged or otherwise unfit bone and replacing it with new matrix. Yet comparative biology and experimental data provide limited support for the theory that matrix renewal is important for maintaining bone strength. Comparatively speaking, BMU remodeling is vigorous in dolphins and whales , the ancestors of which reentered the water some 50,000,000 years ago, yet fish living in the same buoyant environment do not exhibit continuous BMU remodeling. This notable divergence may reflect the ability of fish to extract calcium via gills and scales, whereas marine mammals rely entirely on diet and their skeletons as sources of calcium, making continuous remodeling as valuable to them as it is for terrestrial vertebrates. Regarding experimental data, if matrix renewal was important for the maintenance of bone strength, it might be expected that the inhibition of matrix renewal would have negative impacts on bone biomechanics. However, a clear majority of animal studies indicate that short- and long-term inhibition of remodeling is associated with increases in structural strength and with neutral or positive effects on material (tissue-level) bone strength properties ( Table 31.1 ). These experimental results are the focus of Section 31.13 .
| First author, year | References | Species | Treatment duration | Agent | Structural strength | Material strength |
|---|---|---|---|---|---|---|
| Glatt, 1986 | Mouse | 65 weeks | Pamidronate | ↑ | NR | |
| Toolan, 1992 | OVX rat | 6 months | Alendronate | ↑ | NR | |
| Geusens, 1992 | Monkey | 1 year | Tiludronate | ↑ | NR | |
| Guy, 1993 | Rat | 2 years | Alendronate | ↑ | NR | |
| Balena, 1993 | OVX monkey | 2 years | Alendronate | ↑ | NR | |
| Lafage, 1995 | Minipig | 1 year | Alendronate | ↑ | NR | |
| Lepola, 1996 | Rat | 6 months | Clodronate | ↑ | NR | |
| Peter, 1996 | Dog | 3 years | Alendronate | ↔ | NR | |
| Lalla, 1998 | Rat | 2 years | Ibandronate | ↑ | ↑ | |
| Kippo, 1998 | OVX rat | 6 months | Clodronate | ↑ | NR | |
| Mashiba, 2000 | Dog | 1 year | Risedronate | ↔ | ↔ | |
| Alendronate | ↔ | ↓ | ||||
| Mashiba, 2001 | Dog | 1 year | Risedronate | ↑ | ↔ | |
| Alendronate | ↑ | ↔ | ||||
| ALN/RIS a | NR | ↓ | ||||
| Koivukangas, 2001 | Rat | 32 weeks | Clodronate | ↑ | NR | |
| Itoh, 2002 | OVX monkey | 16 months | Clodronate | ↑ | ↔ | |
| Borah, 2002 | Minipig | 18 months | Risedronate | ↑ | NR | |
| Lees, 2002 | OVX monkey | 2 years | Estrogen | ↔ | ↑ | |
| Raloxifene | ↔ | ↑ | ||||
| Hornby, 2003 | Rat | 1 year | Zoledronic acid | ↑ | NR | |
| Ding, 2003 | Dog | 1 year | ALN/RIS a | ↑ b | NR | |
| Komatsubara, 2003 | Dog | 3 years | Incadronate | ↑ | ↓ | |
| Smith, 2003 | OVX monkey | 16 months | Ibandronate | ↑ | ↔ | |
| Muller, 2004 | OVX monkey | 16 months | Ibandronate | ↑ | ↔ | |
| Glatt, 2004 | OVX rat | 52 weeks | Zoledronic acid | ↑ | NR | |
| Day, 2004 | Dog | 1 year | Risedronate | ↑ | ↔ | |
| Alendronate | ↑ | ↔ | ||||
| Komatsubara, 2004 | Dog | 3 years | Incadronate | ↑ | ↔ | |
| Lees, 2005 | OVX monkey | 2 years | Tibolone | ↑ | ↑ | |
| Estrogen | ↑ | ↑ | ||||
| Li, 2005 | Dog | 1 year | Risedronate | ↑ | ↑ | |
| Atkinson, 2005 | Monkey | 1 year | Denosumab | ↑ | ↔ | |
| Jerome, 2005 | OVX Monkey | 18 months | Cathepsin K inhibitor AAE581 | ↑ | ↑ | |
| Allen, 2006 | Dog | 1 year | Risedronate | ↑ | ↔ | |
| Alendronate | ↑ | ↔ | ||||
| Allen, 2006 | Dog | 1 year | Raloxifene | ↑ | ↑ | |
| Alendronate | ↑ | ↔ | ||||
| Risedronate | ↑ | ↔ | ||||
| Allen, 2007 | Dog | 1 year | Raloxifene | ↔ | ↑ | |
| Allen, 2007 | Dog | 3 years | Alendronate | ↔ | ↓ | |
| Allen, 2007 | Dog | 1 year | Risedronate | NR | ↔ | |
| Alendronate | NR | ↔ | ||||
| Raloxifene | NR | ↑ | ||||
| Ominsky, 2008 | Mouse | 6 months | Denosumab | ↑ | ↑ | |
| Alendronate | ↑ | ↑ | ||||
| Gasser, 2008 | OVX rat | 32 weeks | Zoledronic acid | ↑ | ↑ | |
| Alendronate | ↑ | ↑ | ||||
| Mori, 2008 | OVX monkey | 17 months | Minodronic acid | ↑ | ↔ | |
| Allen, 2008 | Dog | 3 years | Alendronate | ↔ | ↓ | |
| Allen, 2008 | Dog | 1 year | Alendronate | NR | ↓ | |
| Risedronate | NR | ↔ | ||||
| Burr, 2009 | Dog | 3 years | Alendronate | NR | ↔ | |
| Tang, 2009 | Dog | 1 year | Alendronate | NR | ↓ | |
| Risedronate | NR | ↓ | ||||
| Diab, 2009 | Dog | 1–3 years | Alendronate | NR | ↔ | |
| Ominsky, 2011 | OVX monkey | 16 months | Denosumab | ↑ | ↔ | |
| Pennypacker, 2011 | OVX rabbit | 27 weeks | Cathepsin K inhibitor L-006235 | ↑ | ↔ | |
| Alendronate | ↔ | ↔ | ||||
| Masarachia, 2012 | OVX monkey | 21 months | Odanacatib | ↔ | NR | |
| Cusick, 2012 | OVX monkey | 21 months | Odanacatib | ↑ | NR | |
| Yamagami, 2013 | OVX monkey | 17 months | Minodronic acid | ↑ | NR | |
| Kostenuik, 2015 | OVX monkey | 6 months | Denosumab | ↑ | ↔ | |
| 12 months | Denosumab | ↑ | ↔ | |||
| Alendronate | ↔ | ↔ | ||||
| 6-month DMAb then 6-month ALN | ↑ | ↑ | ||||
| Smith, 2015 | OVX monkey | 16 months | Bazedoxifene | ↔ | ↔ | |
| Lee, 2016 | OVX monkey | 16 months | Denosumab | ↑ | ↔ | |
| Duong, 2016 | OVX rabbit | 16 months | Odanacatib | ↑ | ↔ | |
| Alendronate | ↑ | ↔ | ||||
| Allen, 2017 | Dog | 6 months | Alendronate | NR | ↔ | |
| Raloxifene | NR | ↔ | ||||
| Alendronate+raloxifene | NR | ↔ | ||||
| 12 months | Alendronate | ↔ | ↔ | |||
| Raloxifene | ↓ | ↔ | ||||
| Alendronate+raloxifene | ↔ | ↔ | ||||
| Brooks, 2017 | Dog | 12–18 months c | Alendronate | NR | ↔ | |
| Cabal, 2017 | OVX monkey | 20 months | Odanacatib | ↔ | ↔ | |
| Alendronate | ↔ | ↔ |
a Authors combined alendronate and risedronate treatment groups for analysis.
b Size-matched trabecular cores from vertebra were tested without adjusting for bone mass or volume.
c Authors combined 12- and 18-month treatment groups for analysis.
Independent of its matrix-renewing effect, the remodeling trait exhibits other features that could minimize strength losses, even in states where remodeling decreases bone strength. One such feature is the smooth covering of osteoid that is typically deposited on scalloped resorption spaces. Smooth bone surfaces are considered biomechanically optimal in terms of stress distribution , whereas deep scalloped resorption pits ( Fig. 31.2 ) can serve as stress concentrators from which cracks can initiate and propagate . The layer of osteoid deposited via BMU remodeling traditionally follows the smooth contours of the original bone, not that of the jagged surface beneath ( Fig. 31.1 ). This interesting feature explains why the histological identification of remodeling sites versus modeling sites relies on the scalloped appearance of cement lines that characterize the bottom of remodeling spaces, rather than on any distinguishing feature of fluorochrome labels that indicate their refilling. The smoothing effect arising from osteoid deposition at remodeling sites leads to better stress distribution, which confers outsize biomechanical benefits relative to the volume of bone being deposited. Intracortical (i.e., Haversian) remodeling can also be seen to minimize strength losses independent of potential benefits of matrix renewal. The cylindrical pores created by Haversian remodeling negatively impact bone strength , but this porosity frequently accumulates within the endosteal half of the cortex, closest to the central bending axis ( Fig. 31.9 ). This biomechanically favorable distribution of porosity can minimize reductions in long bone bending strength by better preserving moments of inertia .