© Springer International Publishing Switzerland 2015
Jean-Pierre Droz, Bernard Carme, Pierre Couppié, Mathieu Nacher and Catherine Thiéblemont (eds.)Tropical Hemato-Oncology10.1007/978-3-319-18257-5_15Epidemiology of Epstein-Barr Virus and Mechanisms of Carcinogenesis
(1)
Department of Medical Biotechnology, University of Siena, Siena, Italy
(2)
Department of Pathology, University of Siena, Siena, 53100, Italy
Keywords
Epstein-Barr virusLatency programLytic genesmicroRNAsBurkitt lymphoma1 Discovery
The discovery of EBV was a direct consequence of the description of Burkitt lymphoma (BL) in 1958 by Denis Burkitt [6]. The typical geographical distribution of BL led to the idea that environmental factors like Plasmodium falciparum and maybe a virus could have a role in the pathogenesis of the disease. In 1964, Antony Epstein and Yvonne Barr started looking for virus particles in BL biopsies, and their efforts drove to the detection of a herpes virus, which later was called Epstein-Barr virus [19].
2 Structure and Genome of EBV
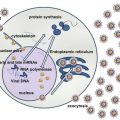
Like other members of the herpes virus family, EBV is an enveloped virus, which contains a core DNA surrounded by an icosahedral nucleocapsid composed of 162 capsomeres [77]. There are three major capsid proteins. The nucleocapsid is in turn enclosed by a protein tegument, which is surrounded by the viral envelope that consists of multiple viral glycoproteins and a tegument [23]. The genome of EBV consists of a 184 kbp double-strand DNA with terminal direct and internal repeats. Upon infection, the virus DNA becomes circular for replication and is maintained as multicopy episome inside the host cell’s nucleus [5]. The EBV genome encodes for a series of almost 100 products interacting with or exhibiting homology to a wide variety of antiapoptotic molecules, cytokines, and signal transducers, hence promoting EBV infection, immortalization, and transformation [14].
3 EBV-Encoded Products
The EBV life cycle includes a lytic phase, resulting in the production of new viral particles, and a latent phase, during which the virus remains silent for the lifetime of the host in memory B cells [61].
3.1 Lytic Proteins
There are more than 90 lytic proteins, which are expressed in a temporally regulated manner [37]. The immediate early proteins, like BZLF1 and BRLF1, are important for regulating the expression of EBV genes and the metabolism of neoplastic cells (i.e., BRLF1) [40]. The early proteins, like DNA polymerase (encoded by BALF5), are important for the replication of the virus’ DNA [32]. Finally, the late proteins, like gp350 and gp110, are the components of the viral particle and are involved in immune evasion [61].
3.2 Latent Proteins
Totally, nine different proteins are expressed by EBV:
(a)
EBNA-1
EBNA-1 is a homodimeric protein which is responsible for EBV DNA replication and persistence by binding to the origin of replication of the EBV genome (oriP). It also plays an important role in transcriptional regulation of the C promoter and contributes to the control of apoptosis, cellular genomic instability, and immune escape by T-cytotoxic response [8, 21, 68].
(b)
EBNA-2
The EBNA-2 protein is localized in the nucleolus and is one of the first viral proteins expressed during EBV infection of primary B lymphocytes. In cooperation with EBNA-LP, EBNA-2 induces the transition of resting B cells from G0 to G1 phase. EBNA-2 is also a key regulator of viral gene and modulates the transcriptional activity of some cellular genes (c-FGR, c-MYC, CD21, CD23, EBI1/BLR2, TNF-α, p55α) [9, 31, 45, 64].
(c)
EBNA-3 family
EBNA-3 family includes EBNA-3A, EBNA-3B, and EBNA-3C, also known as EBNA-3, EBNA-4, and EBNA-6, which are present in tandem in the viral genome and are indispensable for B-cell transformation by the virus. A large number of target genes of EBNA-3 family have been identified, including BCL6, IRF-4, BLIMP-1 (involved in B-cell differentiation), NFATC2, BACH2, EBF1CD21, CD40, CHK2 (disrupting G2/M checkpoint of cell cycle), RAC1, LYN, TNF-α, JAK-STAT, and MAPK signaling pathway members [26, 47, 79, 88].
(d)
EBNA-LP
EBNA-LP, also known as EBNA-5, is the first viral latent protein to be expressed after the infection of naive B cells along with EBNA-2. EBNA-LP has been shown to increase the transcription effect of EBNA-2 in activating the expression of LMP-1 and LMP-2 and in inducing cyclin D2, and thus causing the transition of G0/G1 checkpoint. EBNA-LP has been shown to bind to p53, Rb, heat shock protein 70, DNA protein kinase catalytic subunit, HA95 (a nuclear protein that is involved in mitosis), and α- and β-tubulin [35, 54].
(e)
LMP-1
LMP-1 consists of a short cytoplasmic N-terminus, six transmembrane domains that are located in lipid rafts on the membrane of the cell, and a long cytoplasmic C-terminus. The protein is a functional analog of a constitutively activated form of CD40, a receptor which belongs to the TNF receptor family, with an important role in B-cell activation. By this means, LMP-1 can activate important signaling pathways (NF-κB, MAPK, interferon-regulatory factor 7 and PI3K). LMP-1 has also an antiapoptotic effect achieved by regulating the expression of different cellular proteins like MYC, BCL2A1, TNFAIP3, and CL2 [39, 41, 58, 73, 89].
(f)
LMP-2 family
LMP-2 family includes two forms of LMP-2 proteins (LMP-2A and LMP-2B) which are necessary for B-cell transformation and B-cell receptor activation. LMP-2A has been described as a B-cell receptor (BCR) signaling mimic inducing downstream pathways that inhibit apoptosis and promote cell survival [86]. Moreover, LMP-2A cooperates in reprogramming normal B-lymphocyte functions and enhances MYC-driven lymphomagenesis [17, 20].
4 RNA Molecules
EBV expresses two small RNA known as EBV-encoded RNAs (EBER1 and EBER2) and several microRNAs (miRNAs) [61].
(a)
EBER family
EBERs are the most abundant viral transcripts producing during latent infection by EBV. Both of them are almost 170 nucleotide long and are expressed in all EBV latencies and in all the EBV-infected cells in vivo. Their high level of expression (almost 106 per cell) rendered them as invaluable targets in diagnostics of EBV infection. The evolutionary conservation of EBERs and their ubiquitous expression suggest that they may play a key role in EBV biology. In fact, they have been demonstrated to elicit a variety of effects on cell growth, apoptosis, protection from protein translation shutoff, induction of cytokines, and lymphomagenesis [78].
(b)
MicroRNAs
The miRNAs are small noncoding single-strand RNA molecules (~22 nucleotide long) which can regulate the expression of their target genes at posttranslational level [13, 25, 42, 50]. EBV expresses 44 mature miRNAs from 25 precursors, which are mapped in two regions of the EBV genome: BHRF1 (Bam HI fragment H rightward open reading frame I) and BART (Bam HI-A region rightward transcript), from which three belong to the BHRF family and the rest to the BART family. Expression of EBV miRNAs is dependent on a variety of factors including the host cell type. Viral miRNAs regulate both cellular and viral genes and thus play important roles in maintaining the EBV latency. In addition, they provide a potent mechanism for the virus to modulate the cellular environment by regulating host cell growth, survival, apoptosis, and immune evasion. EBV is also able by itself to affect expression of cellular miRNAs, thereby regulating cellular gene expression to enhance EBV effects in the pathogenesis of viral-associated diseases [4, 7, 44, 51, 56, 65].
5 Persistent Infection, Latency, and Viral Reactivation
In the generally accepted model of EBV persistence, the virus initiates infection by crossing the epithelium of the oropharynx and infecting resting naive B cells in Waldeyer ring, the major receptor for the virus on B cells being CD21 [57]. Although B cells represent the principle targets of EBV, epithelial cells are important for lytic infection, producing viral progeny that amplifies cell-to-cell spreading and enables transmission to the host. After the infection, EBV establishes persistent infection characterized by the sequential employment of a series of latency transcription programs that allow the virus to drive the newly infected naive B cell into the memory B-cell compartment [38, 62, 75, 76]. In this setting, the virus maintains a latent state as an episome and expresses no viral genes as viral DNA becomes methylated over time. Specifically, upon infection, EBV delivers its linear genomic DNA that is epigenetically naive to the nucleus of infected cells where it forms a circular plasmid and initiates a phase in the viral life cycle termed “pre-latent” [34]. This phase is characterized by the co-expression of two distinct sets of viral genes, latent and lytic. The expression of EBNAs, LMPs, EBERs, and miRNAs activates the quiescent B lymphocytes, which become lymphoblasts and begin to proliferate. At this early stage, the concomitant expression of certain lytic genes, which encompass transcription factors and cytokines, protects the activated B lymphocytes from endogenous stress, immediate activation-induced apoptosis, and, presumably, DNA damage response signals. The pre-latent phase is transient and ceases within 1–2 weeks; during this phase, histones acquire substantial epigenetic modifications over time, and the viral DNA becomes extensively methylated at CpGs. The pre-latent phase is replaced by a strictly latent phase, in which the virus establishes a stringent and stable virus-host relationship. Viral gene expression is entirely restricted to EBNAs, LMPs, EBERs, and BHRF1 miRNAs (latency III). This program (named growth transcription program) may be important for cancer development, because it is capable of initiating the activation of B cells in vitro into continuously proliferating lymphoblastoid cell lines (LCL). Since the activated naive B lymphoblasts in vivo are targeted by the immune system, EBV adopts a strategy to survive in the organism. In fact, infected B cells rapidly migrate to the follicle to participate in a germinal center (GC) reaction and continue to proliferate by switching to a more restricted latency program (latency II) where only EBNA-1, LMP-1, LMP-2 s, EBERs, and BART miRNAs are expressed. This leads to the removal of the differentiation block imposed by EBNA-2 protein (default transcription program). Ultimately, the cells leave the germinal center as resting memory B cells at the site of long-term latent persistent infection. In memory B cells, all viral protein expression is extinguished (latency 0) except when the cells divide and express EBNA1 (the protein required for replication of the viral genome), EBERs, and BART miRNAs (latency I). This mechanism is thought to allow EBV-infected cells to remain hidden from the immune system, enabling lifelong persistence. Upon receiving certain activation signals (BCR stimulation, hypoxia, TGF-β, DNA damage, and chemical agents), latency can be disrupted, and the virus reactivates, switching to a lytic phase, with subsequent lysis/death of infected cells and release of virions that infect more cells and enable virus transmission also from host to host [37]. Lytic phase is also able to enhance tumor growth through growth factors and immunosuppressive cytokines.
6 Epidemiology and EBV Infection
The International Agency for Research on Cancer has demonstrated that more than 90 % of adults worldwide are infected with EBV [27, 72]. The age at primary infection varies substantially worldwide, and exposure to EBV is likely to be due to socioeconomic factors. In developed countries, two peaks of infection are seen: the first in very young preschool children aging 1–6 years and the second in adolescents and young adults aging 14–20 years [12]. In developing countries, infection occurs at a much earlier age so that 90 % of children over the age of 2 are seropositive [27]. There is no consistent difference in EBV seroprevalence by sex. EBV transmission occurs through saliva; however, in developing countries, infection is acquired mainly for crowding and/or the practice of pre-chewing food for infants, whereas in developed world, transmission is more likely because of intimate oral exposure (the so-called kissing disease). Primary EBV infection takes place in the oral route to which the virus is conveyed by saliva droplets from infected individuals [66]. The nature of the target cells in the oral mucosa is still controversial, but there is agreement that B cells are infected at some stage of the process as they traffic in close proximity to oropharyngeal epithelium. If infection is delayed to adolescence or adulthood, it can cause an infectious mononucleosis syndrome, characterized by the polyclonal expansion of infected B cells. The disease is self-resolving as it elicits a strong cellular immune response which brings the infection under control, and newly infected cells are efficiently removed by the cytotoxic T-cell response [15]. Following resolution of primary infection, the virus establishes a lifelong persistence in memory B cells that usually remains clinically silent. In this B-cell reservoir, viral expression is entirely repressed; this is how these infected cells can persist in the face of a competent immune system [16].
7 EBV-Associated Malignancies
The strategy of EBV is to persist in healthy chronic carriers, avoid killing the cell, and prevent the cell from becoming a target for destruction by the immune system [24]. Therefore, the virus initiates an ongoing, tightly orchestrated interplay between itself, the host B cell, and the immune system that allows EBV to persist and eventually activate cellular growth control pathways. To promote viral persistence, EBV has evolved a number of strategies to modulate the host-immune response, including inhibition of immune cell functions and of apoptosis and interfering with antigen processing and presentation pathways [74]. However, in this long-standing interaction, something wrong could occur, leading to dysregulation of cellular pathways or perturbation of host immunity, thus resulting in the development of EBV-associated malignancies. In immunodeficiency settings, for the absence of an effective T-cell surveillance, latently infected cells in the peripheral blood or persistently infected cells on the oropharynx increase in number and usually express all the viral genes (growth program—latency III), thus activating multiple intersecting cellular pathways and producing its own miRNAs that alter the host cell regulatory machinery [28]. However, in immunocompromised patients, lymphomas other than posttransplant lymphoproliferative disorders show more restricted forms of latent gene expression, reflecting a complex pathogenesis that may involve additional cofactors. These tumors might evolve from EBV-transformed LCL-like cells through the acquisition of additional cellular genetic changes that render certain viral functions redundant. In immunocompetent individuals, EBV-induced cancerogenesis is a multistep process where oncogenic effect of EBV products in association with additional genetic, environmental (i.e., viral infection by HHV8, CMV, arbovirus, and HIV) [60], and microenvironmental factors (i.e., Euphorbia tirucalli) [46] contributes. There is increasing evidence that the microenvironment of the EBV-infected B cell can regulate virus gene expression and modulate the function of individual virus proteins [84]. For example, the cytokines interleukin-21 (IL-21) and IL-2, along with intercellular interactions such as CD40 ligation, all present in the germinal center of the tonsil, have been shown to downregulate the expression of EBNA-2 and upregulate the expression of LMP-1, thus imposing a type II expression profile similar to that observed in Hodgkin lymphoma [84]. Changes to the microenvironment of the infected B cell might also help to explain the double function of LMP-1 in asymptomatic host: drive the differentiation of EBV-infected GC B cells and elicit a potential oncogenic effect [84]. Moreover, as the healthy immune system tends to remove EBV-infected cells expressing multiple immunogenic viral proteins, there is a selective negative pressure for which EBV-induced tumors evolve in a way that is EBV-independent by switching off EBV genes and by acquiring compensating genetic alterations to survival and growth [82, 83]. In addition, the view that only the latent phase of viral gene expression is important during the development of EBV-associated malignancies has recently changed, suggesting a potential role also for viral gene products [3]. There are several potential mechanisms by which EBV lytic gene expression could contribute to the growth of EBV-associated tumors in vivo: by increasing the horizontal transmission of the virus from cell to cell, lytic infection may increase the total number of latently infected cells; moreover, viral lytic genes, or cellular genes induced by viral lytic proteins, could potentially encode paracrine factors and angiogenic factors that promote tumor growth and immune escape [29, 33].
EBV-associated malignancies include B- and T-/NK-cell lymphomas as well as non-hematological malignancies [59, 71, 74, 80]. In particular, EBV-positive B-cell lymphomas might be thought of as rare accidents of EBV colonization of B cells, and the pattern of virus latency observed in the different histotypes might be taken as evidence of the stage of B-cell differentiation from which the tumor is derived [85].
Lymphoma:
Hodgkin lymphoma both in non-immunodeficient and in immunodeficient (congenital, HIV, posttransplant) patients
Non-Hodgkin B-cell lymphomas (NHL) both in non-immunodeficient (BL, diffuse large B-cell lymphoma (DLBCL) of the elderly, pyothorax-associated lymphoma (PAL), and lymphomatoid granulomatosis (LYG)) and in immunodeficient patients (BL, primary central nervous system DLBCL, primary effusion lymphoma (PEL), plasmablastic lymphoma (PBL), posttransplant lymphoproliferative disorders (PTLD))
Non-Hodgkin NK-cell or T-cell lymphomas: peripheral T-cell lymphomas, angioimmunoblastic T-cell lymphoma, extranodal nasal-type NK-/T-cell lymphoma, T-cell lymphoproliferative disorders of the childhood, and EBV-associated cutaneous T-cell lymphoproliferative disorders
Carcinoma:
Nasopharyngeal
Gastric
Lymphoepithelioma-like
Sarcoma: in immunodeficiency setting (leiomyosarcoma)
8 EBV-Associated Lymphomas
Hodgkin lymphoma (HL) is a neoplasia composed of tumor cells designated Hodgkin cells or Reed-Sternberg cells residing in an abundant heterogenous admixture of nonneoplastic inflammatory cells. The tumor cells are usually ringed by T cells in a rosette-like manner. EBV positivity in lymphoma tissue is detected in 70 % of mixed cellularity, 95 % of lymphocyte depleted, and 10–40 % of nodular sclerosis; the lymphocyte-predominant subtype is almost always EBV negative. The role that EBV plays in HL is still not fully understood. EBV gene expression follows the latency II pattern with EBNA-1, LMP-1, LMP-2A, and LMP-2B and the EBERs being expressed. The role of EBNA-1 in carcinogenesis and the oncogenic capabilities of LMP-1, LMP-2A, and LMP-2B and the EBERs have been addressed above. In addition, EBV provides important antiapoptotic signals that prevent cell death in HL progenitors lacking a functional BCR. Loss of BCR and of other key components of the BCR signaling machinery could be important for the pathogenesis because they might protect HL progenitors from entry into the EBV-replicative cycle and subsequent cell death. In addition, chronic inflammation in the microenvironment might not only dictate the pattern of EBV gene expression but also modulate the oncogenic functions of individual EBV genes such as LMP-1 [48].
Burkitt lymphoma (BL) is a highly aggressive B-cell non-Hodgkin lymphoma characterized by peculiar clinical, morphological, immunophenotypical, cytogenetic, and gene expression profile features. Differences in geographical distribution and association with EBV and HIV account for three epidemiologically distinct variants: the endemic BL (eBL), sporadic BL (sBL), and immunodeficiency-associated subtypes. EBV has been detected in virtually all cases of endemic variant, 15–20 % of the sporadic form and 30–40 % of the immunodeficiency-related BL. Most EBV-positive cases exhibit the latency I type; however, we have recently demonstrated a noncanonical latency program in BL characterized by the expression of either LMP-1 and LMP-2 along with some lytic gene products (Leoncini L et al., 2014, personal communication). EBV contributes to the pathogenesis of BL by providing the antiapoptotic signals necessary to override MYC-induced cell death, thanks to EBNA-1 expression. Since only one EBNA-1 is expressed in BL, a substantial role for EBV-encoded miRNAs in BL does exist in regulating apoptosis, gene expression, immune system, and other processes [22, 51, 53, 63].
Posttransplant lymphoproliferative disorders (PTLD) is a clinicopathological entity encompassing a heterogeneous group of disorders that follow solid-organ transplant or bone marrow allograft as a consequence of immunosuppression and range from reactive hyperplasia to malignant monoclonal forms. According to the WHO classification, they are divided into early lesions (reactive plasmacytic hyperplasia and mononucleosis-like syndrome), polymorphic lesions, monomorphic lesions, and Hodgkin-like lesions. EBV has been linked to most PTLDs, with a near 100 % association in the early occurring cases (within a year) and in PTLD-associated HL. The EBV-negative PTLDs constitute approximately 20 % of all cases, have a tendency to late occurrence (>5 years posttransplant), and have an unknown etiology. Type III latency is exhibited by the EBV-positive B cells in PTLD, although some studies have reported a more restricted latency pattern. The wide expression of the latent EBV-encoded proteins strongly suggests an important role that EBV may play in the oncogenic process. Because approximately 50 % of PTLD cases are derived from GC B cells lacking a functional BCR for crippling mutations and because these cells manage to escape apoptosis despite lacking antigen affinity, it is believed that EBV aids in rescuing these cells from an imminent programmed cell death. As in HL, LMP-1 and LMP-2A may replace survival signals induced by activated BCR and CD40 receptors and also activate the NF-κB signaling pathway, inducing proliferation of neoplastic cells. The decreased cytotoxic T-cell surveillance because of immunosuppression in PTLD patients is also believed to greatly facilitate the actions of EBV [43].
Lymphomatoid granulomatosis (LYG) is a rare angiocentric and angiodestructive B-cell lymphoproliferative disorder that is composed predominantly of reactive T cells and fewer neoplastic EBV-positive B cells. The most common site of involvement is the lung, with less frequent involvement of other extranodal sites such as the skin, kidney, liver, and central nervous system. The latency pattern for EBV has not been extensively studied in lymphomatoid granulomatosis; but one group has reported the detection of LMP-1 and EBNA-2 by immunohistochemistry and the EBERs by in situ hybridization, which is indicative for type III latency. The near 100 % EBV association with lymphomatoid granulomatosis and the presumed wide expression of EBV latent-encoded proteins strongly infer that EBV is not just an innocent bystander but may play a crucial role in the pathogenesis of the disease [18].
Other B-cell lymphomas: PAL is an NHL developing in the pleural cavity after a long-standing history of pyothorax; it is characterized by a type III latency [70]. EBV-positive DLBCL of the elderly is a clonal B-cell lymphoid proliferation that occurs in patients over the age of 50 years and without any known immunodeficiency or prior lymphoma, showing a type II–III latency [36]. PEL, a rare and distinct tumor that affects body cavities without a detectable tumor mass, is predominantly associated with HHV-8 infection. The expression of EBV-encoded proteins is consistent with type II latency [10]. PBL is characterized by a diffuse proliferation of large neoplastic cells, most of which resemble B immunoblasts, but in which all tumor cells have the immunophenotype of plasma cells. The exact role elicited by EBV in inducing lymphomagenesis still remains unknown as well as its latency type, although a type I latency has been suggested [49]. However, we have recently performed an miRNA’s profiling and a complete study of EBV products, finding the expression of EBV-encoded miRNAs belonging to the BART locus and a noncanonical latency of the virus with an abortive lytic cycle [3]. Lastly, virtually 100 % of primary central nervous system DLBCL shows EBV association with a latency III type [69].Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree