Fig. 1
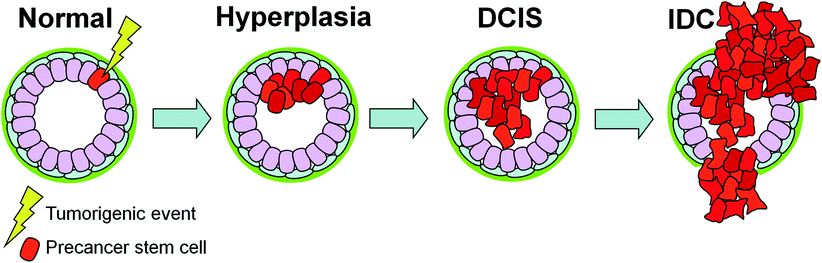
The stages of breast cancer. Within the normal mammary duct and TDLU, an inner layer of luminal epithelial cells and a basal layer of myoepithelial cells are surrounded by a basement membrane. In benign hyperplasia, epithelial cells have proliferated within the duct while retaining a largely normal phenotype. In ductal carcinoma in situ (DCIS), the cells appear phenotypically different from normal epithelia—poorly differentiated DCIS more so than well-differentiated DCIS—yet are still confined by the basement membrane. DCIS is classified into several subtypes based on its histopathological presentation, including grade, growth pattern, and necrosis. Once the neoplastic cells invade beyond a degraded basement membrane into the surrounding breast stroma, they have transitioned to invasive ductal carcinoma (IDC). In subsequent figures, the luminal cells, myoepithelial cells, and basement membranes are displayed with the same color scheme as shown above
Before the widespread use of screening mammography, DCIS comprised only ~1–2 % of all newly diagnosed breast cancers (Allred 2010). These DCIS lesions were large, palpable, and described as “comedo” due to the necrotic ooze that poured out of them when squeezed. With the advent of screening mammography, the incidence of DCIS skyrocketed to comprising 20–30 % of newly diagnosed breast cancers in the US today (Kerlikowske 2010; Virnig et al. 2010). Mammographic screening also revealed a previously unexplored spectrum of DCIS lesions with diverse histological, molecular, and genetic characteristics. In this chapter, we will delve into what is known regarding the biology of this remarkably heterogeneous disease, and how it transforms itself into invasive carcinomas.
Radiographically, the classic presentation of DCIS comes in the form of calcifications on X-ray mammography, with a diversity of shapes and spatial distributions (D’Orsi 2010). Magnetic resonance imaging (MRI), while not as widely used X-ray mammography, has the highest sensitivity for DCIS of all clinically used imaging modalities (Kuhl et al. 2007). DCIS presents with a distinctive nonmass-like morphology on MRI, with clumped internal enhancement in a segmental, linear, or regional distribution (Jansen et al. 2007; Lehman 2010; Jansen 2011). Although DCIS is a preinvasive cancer, it is not necessarily small—the average size of DCIS is 10–20 mm (Allegra et al. 2010), but some lesions can extend through most of a major breast lobe (Tot 2010).
Histologically, DCIS is classified according to metrics initially developed for IDC such as grade. DCIS is categorized as low, intermediate, or high grade based on a variety of nuclear features including size and mitotic activity. Overall, DCIS lesions are classified either as well-, moderately-, or poorly-differentiated depending on how closely they resemble normal cells (Fig. 1) (Allred 2010). Unlike IDC, DCIS has distinctive growth/architectural patterns describing how the cells are distributed within the duct lumen: solid, papillary, micropapillary, cribriform or comedo. Necrosis is extensive and predominant in comedo DCIS while focal or absent in the other architectural subtypes.
Current treatment paradigms for DCIS include mastectomy (Hwang 2010), lumpectomy alone, or lumpectomy with radiation therapy (Kane et al. 2010). Approximately 20 % of DCIS patients treated with lumpectomy alone experience a recurrence, half as DCIS and half as invasive cancer (Kerlikowske et al. 2010). Both radiation therapy and adjuvant Tamoxifen have been shown to reduce recurrence rates by approximately 50 % in DCIS (Correa et al. 2010; Eng-Wong et al. 2010; Solin 2010). Given current management strategies, the prognosis for DCIS is excellent with a 10-year disease-specific survival rates of 98 % (Bijker and van Tienhoven 2010; Kerlikowske et al. 2010; Schnitt 2010). Concomitantly high incidence and survival rates imply that by 2020, an estimated 1 million women will be living with a diagnosis of DCIS in the US alone (Allegra et al. 2010).
In many ways, DCIS represents a success story in breast cancer management—the combination of early detection and effective treatments yields excellent cure rates in these patients. However, there is a growing clinical concern that a significant portion of DCIS patients are actually being over-diagnosed and over-treated, as indirect evidence suggests that approximately 50 % of DCIS will not progress to invasive, life-threatening diseases (Erbas et al. 2006; Kuerer et al. 2009; Allegra et al. 2010). However, there is no way now to identify this subset of women with indolent disease who may benefit from less aggressive therapeutic interventions. As highlighted by a 2009 National Institutes of Health State of the Science Conference Statement, the primary task for research going forward is to identify subsets of DCIS patients based on their risk of progression to invasive carcinoma (Allegra et al. 2010). An important step toward achieving this goal is to better characterize the biology of DCIS and understand the key events responsible for its transition to invasive carcinoma.
2 The Biology of DCIS
Cancer is a genetic disease wherein malignant transformation is driven by changes in DNA. There are many types of alterations that can occur, including gross chromosomal gains/losses, genomic amplification, genomic rearrangements, activation of oncogenes, and inactivation of tumor suppressor genes via mutation, deletion, methylation, or transcriptional repression. As a consequence of these genomic events, the cancer transcriptome can exhibit marked irregularities compared to normal tissue. These transcriptional changes, usually represented by up or downregulated gene signatures in gene-expression profiles, are then translated into the proteome, resulting in proteins that are overexpressed, lost, or have amino-acid substitutions that confer gain or loss of function. Additionally, post-transcriptional and post-translational modifications not reflected at the mRNA level can also affect the function, stability, and distribution of proteins. Characterization of cancer at all levels—DNA, RNA, and protein—is a major goal of cancer biology, as it can elucidate both the molecular pathways critical for cancer etiology and identify subtypes of disease that provide prognostic information or predict therapeutic efficacy. For example, breast cancers that express estrogen receptor (ER) are often treated with selective ER modulators in an adjuvant setting.
In the larger context of breast cancer biology and evolution, DCIS is thought to be a precursor to IDC. However, it is not direct observation of DCIS progression to invasive disease in women that underpins this belief—due to the obligate surgical excision of newly diagnosed cancers including DCIS it is very challenging to track the natural history of disease in women (Erbas et al. 2006). Rather, the status of DCIS as a precursor to IDC is due to a large body of compelling yet indirect evidence demonstrating connections between the two. This evidence includes sharing similar risk factors (Kerlikowske 2010), the increased risk of recurrence as IDC at the tumor bed for women with DCIS, frequent co-existence of DCIS and IDC together in the same lesion (Allred et al. 2008), and the finding that DCIS and IDC harbor many of the same molecular and genetic abnormalities (Polyak 2010). Further evidence temporally linking DCIS to IDC comes from follow-up of women with DCIS initially misdiagnosed as benign disease showing that 14–50 % eventually develop IDC (Erbas et al. 2006), and work in animal models where more direct observations of preneoplastic progression are possible (Maglione et al. 2001, 2004; Namba et al. 2004, 2006; Damonte et al. 2008). Although our primary focus is on DCIS, we will begin with a brief examination of the key biological features of invasive cancers. This will serve as a platform for our subsequent discussion of the genomic, gene-expression, and protein level characteristics of DCIS.
There is a caveat to what is presented below relating to the challenge of applying molecular assays to DCIS surgical specimens. Limitations include small patient cohorts, mixed screening and unscreened populations, and small lesion size. An additional challenge is getting access to tissue in the first place, particularly for patients with pure DCIS where most of the tissue must be processed to histologically rule out microscopic regions of invasion. Consequently, many studies describing the molecular features of DCIS often include lesions that have both IDC and DCIS components. The unavoidable limitations of studies of DCIS are important to consider when interpreting the empirical data.
2.1 The Genome and Transcriptome of Invasive Carcinoma
On the genomic level, grade I invasive carcinomas show low levels of genomic instability, with frequent recurrent loss of 16q and gain at 1q. Conversely, grade III invasive cancers show higher levels of genomic instability with amplification of 17q12, 11q13, loss of 8p, 11q, 13q, and gains of 1q, 8q, 17q, and 20q (Buerger et al. 1999a, b; Roylance et al. 1999). Grade II IDC can exhibit mixed features between these two (Sgroi 2010). The frequent loss of 16q in low but not high-grade carcinomas is salient for modeling breast cancer evolution. It suggests that the majority of grade III invasive carcinomas do not arise from a grade I precursor, as this would imply the recovery of genetic material at 16q. Rather, the restriction of the loss of 16q to low-grade invasive carcinoma suggests that the different grades of invasive carcinoma arise from different pathways. Empirical data complicate this interpretation since approximately 20 % of grade III IDC also harbor loss of 16q (Roylance et al. 1999), suggesting that the low to high-grade transition may occur in a subset of women. These high-grade lesions with loss of 16q are mostly ER+ (Smart et al. 2011).
Comprehensive gene expression profiling based on the influential studies by Perou et al. (2000) and Sorlie et al. (2001) have resulted in the molecular subtype classification of breast cancers into four categories. The major distinction is at the level of ER, and secondarily on the level of human epidermal growth factor receptor 2 (HER2). Two subtypes are ER–: the basal (HER2–) and ERBB2 (HER2+) groups, and two subtypes are ER+: the luminal A (HER2–) and luminal B (HER2+) groups. These transcriptome-based subtypes correlate well with other histological and clinical features; luminal A and B often exhibit lower grade and more favorable prognosis compared to the basal and ERBB2 subtypes, which are often higher grade with poorer prognosis (Sorlie et al. 2001).
2.2 The Genome and Transcriptome of DCIS
Remarkably, there are essentially no progression-specific changes in DCIS compared to IDC on the genome or transcriptome level. On the contrary, the characteristics of DCIS mirror those of invasive breast cancer, including exhibiting distinct molecular characteristics by grade (Porter et al. 2001, 2003). Low-grade DCIS also harbors frequent recurrent loss of 16q, while high-grade DCIS shows a more complex genomic pattern with loss at 8p, 11q, 14q, gains at 1q, 8q, and 17q and high level of amplification at 17q12 and 11q13 (Buerger et al. 1999a, b; Hwang et al. 2004; Vincent-Salomon et al. 2008). DCIS can also be classified into the four molecular subtypes of breast cancer (basal, ERBB2, luminal A and B) that in turn correlate with different grades (Vincent-Salomon et al. 2008). This evidence suggests that the bulk of genetic transformation in breast cancer does not occur during the (putative) DCIS to IDC phase change. Indeed, Ma et al. performed gene-expression profiling comparisons of normal breast tissue, DCIS, and IDC lesions and found that the largest transcriptional changes occur from the normal to DCIS transition, not the DCIS to invasive transition (Ma et al. 2003). With these qualitative similarities between DCIS and IDC noted, it is important to point out that quantitative analysis does reveal differences, for example in the levels of gene expression (Ma et al. 2003) and in the proportions of molecular subtypes found (Vincent-Salomon et al. 2008; Clark et al. 2011; Yu et al. 2011). Therefore, while similar genes may be affected in DCIS versus IDC, there may be differences in the dynamics (i.e., timing and levels) of such transcription.
Thus, from a genome and transcriptome perspective, we have not found molecular alterations that are specific to DCIS and not invasive disease. Rather there are dramatic changes from normal tissue to DCIS, and also between different grades regardless of whether invasive or noninvasive (Ma et al. 2003). This evidence further supports a multi-pathway grade-specific model of breast cancer progression that extends to the preinvasive stage.
2.3 Molecular Markers of DCIS
Analysis of protein expression in DCIS, most often assessed via immunohistochemistry (IHC) on tissue sections, is an important tool for studying the molecular pathways altered in DCIS and identifying subtypes of disease based on differential protein expression. An advantage of IHC is that the spatial distribution of proteins, both within the cell (i.e., cytoplasmic or nuclear) and across the lesion, can be visualized. An inherent limitation is that for many molecular markers, methods for determining positivity in DCIS have not been fully standardized. Most commonly, a lesion is classified as positive (‘+’) if a certain threshold number of cells within the lesion exhibit positive staining, and negative (‘−’) if that threshold is not met (Lari and Kuerer 2011). The most studied molecular markers in DCIS are ER and HER2. However, other important signaling pathways responsible for regulating proliferation, apoptosis, angiogenesis, and genome stability have also been examined (Lari and Kuerer 2011) (Table 1).
Table 1
Summary of the molecular markers used to characterize DCIS
Molecular markers | Functions | Molecular signatures correlating with increased risk of recurrence |
|---|---|---|
ER, PR | Steroid receptors | ER− HER2+ ER−/HER2+ ER−/HER2 +/Ki-67+ |
HER2 | Regulates proliferation and apoptosis | |
p53 | Regulates cell-cycle, apoptosis, and genomic stability; p53 is an important tumor suppressor | p53+ |
Rb/p16 pathway | Regulates cell-cycle; Rb is an important tumor suppressor | p16+ Ki-67+ COX-2+ p16+/COX-2-/Ki-67+ (DCIS recurrence) p16+/COX-2+/Ki-67+ (invasive recurrence) |
Ki-67 | Proliferation marker | |
COX-2 | Enzyme for prostaglandin synthesis; expressed during inflammatory response | |
Akt/PTEN pathway | Regulates proliferation, survival and motility; PTEN is an important tumor suppressor | |
BRCA1/2 | DNA damage repair | |
c-myc | Transcription factor that can activate proliferation; c-myc is a proto-oncogene | |
VEGF, vascular patterns | Angiogenesis and vascular markers | |
Cyclin A, cyclin E, p21, p27 | Cell-cycle regulators | p21+ |
Bcl-2, Bax, Survivin | Apoptosis regulators | Bcl-2− Survivin+ |
Estrogen and progesterone receptors. Approximately 70–80 % of DCIS lesions are ER+ (Tamimi et al. 2008; Kerlikowske et al. 2010). ER+ DCIS is less likely to be high grade, HER2+, p53+, and Ki-67+ or display comedo growth patterns (Lari and Kuerer 2011). PR expression correlates closely with ER.
HER2. HER2 is a member of the ErbB family of proteins and is involved in regulating cellular proliferation and apoptosis through a variety of signaling pathways, including MAPK, Akt, and STAT. Approximately 20–30 % of DCIS patients have HER2 overexpression (Tamimi et al. 2008; Kerlikowske et al. 2010), as assessed by IHC or fluorescence in situ hybridization (FISH) to measure amplification of the ErbB2 gene. HER2+ DCIS is more likely to be ER−, PR−, Bcl-2−, p53+, Ki-67+, p21+, have high nuclear grade and exhibit comedo necrosis (Lari and Kuerer 2011). Although the proportion of HER2+ DCIS is comparable to IDC, the triple-negative i.e., ER−/PR−/HER2− phenotype is less common in DCIS (Kuerer et al. 2009)
p53. p53 is an important tumor suppressor protein that plays a critical role in a cell-cycle regulation, apoptosis, and genomic stability. Mutations in the TP53 gene occur in approximately 20 % of invasive breast cancers and 10–30 % of DCIS (Livasy et al. 2007; Kerlikowske et al. 2010) and can manifest itself on IHC as accumulated p53 protein expression. DCIS lesions that are p53+ are more likely to have high nuclear grade and comedo necrosis (Lari and Kuerer 2011). Likewise, TP53 mutations are more frequently observed in basal-like and HER2+ DCIS than those in the luminal group (Polyak 2010). As discussed in more detail below, p53 expression is correlated with increased intratumor heterogeneity in DCIS, which may arise as a result of increased genomic instability upon aberrant p53 activity.
Rb/p16 pathway. A key regulator of cellular proliferation is the Rb tumor suppressor pathway. Rb functions to inhibit expression of genes required for cellular proliferation by assembling transcription repression complexes. This activity is reduced in proliferating cells by the action of CDK/cyclin complexes that phosphorylate Rb. In particular, the CDK4/cyclin D complex is the rate-limiting step for Rb inactivation; this complex is itself counteracted by p16. The Rb-cyclinD-p16 network defines a pathway that is functionally inactivated in most cancers, although Rb itself may not be mutated or lost. For example, simultaneous expression of both p16 and Ki-67 could be indicative of Rb functional loss, as recently suggested by Witkiewicz et al. for DCIS (Witkiewicz et al. 2011). Abrogation of the Rb pathway has recently been proposed as a mechanism by which DCIS can survive the hypoxic and stressful intraductal environment (Espina and Liotta 2011). Approximately 70, 60, and 35 % of DCIS lesions are Rb+, cyclin D1+, and p16+, respectively (Gauthier et al. 2007; Millar et al. 2007; Kulkarni et al. 2008; Okumura et al. 2008; Kerlikowske et al. 2010), while 22 % are both p16+/Ki-67+ (Kerlikowske et al. 2010).
Akt/PTEN pathway. The Akt pathway regulates many diverse biological functions, including cellular motility, proliferation, and survival. Components of this pathway, particularly PTEN, are deregulated in a wide spectrum of human cancers. The PTEN tumor suppressor is an important negative regulator of the Akt pathway, and is altered in approximately 40 % of invasive breast cancers. Using tissue microarrays comprised of pure DCIS and IDC, Bose et al. evaluated the expression of Akt pathway members at different stages of breast cancer progression. They found that Akt and its downstream proteins mTor and S6 are activated in 30 % of DCIS and IDC lesions. Interestingly, PTEN was differentially expressed by stage: only 11 % of DCIS exhibited loss of PTEN compared to 26 % of invasive cancers (Bose et al. 2006). PTEN loss has also been linked to the basal subtype of DCIS (Polyak 2010).
BRCA1/2. The tumor suppressors BRCA1 and BRCA2 function to repair damaged DNA. Women with deleterious mutations in the BRCA1/2 genes are at high risk for developing invasive breast cancers with earlier onset. A prior misconception had been that the natural history of BRCA-related tumorigenesis differed significantly from its sporadic counterpart, by passing very quickly through the in situ phase (if at all). However, more recent studies have reaffirmed DCIS as a relevant stage in BRCA-related breast cancer. In BRCA mutation carriers, DCIS is diagnosed in comparable proportion as in other patient cohorts (Hwang et al. 2007; Arun et al. 2009). Furthermore, the prevalence of BRCA1 and BRCA2 mutations among DCIS lesions is comparable to that of invasive carcinomas (Claus et al. 2005; Smith et al. 2007). Interestingly, recent evidence suggests that DCIS is more difficult to detect with MRI in BRCA1 mutation carriers compared to other high-risk women including BRCA2 mutation carriers (Sardanelli et al. 2010; Jansen 2011; Warner et al. 2011). This points to possible differences in underlying physical or physiological properties, leading to evasion of MRI detection.
c–myc. The proto-oncogene c-myc is a transcription factor that plays a role in proliferation, malignant transformation, and apoptosis. The observed genomic alterations in DCIS implicate the activation of the c-myc gene located at 8q24; this includes frequent observation in DCIS of gains at 8q and the loss of CTCF, a transcriptional regulator of c-myc, at 16q. Approximately 60 % of DCIS are c-myc+, and these lesions are more likely to be HER2+ and proliferative (Altintas et al. 2009).
Angiogenesis. As a non-invasive stage of breast cancer, DCIS is not necessarily associated with dense, leaky neovasculature as invasive cancers can be. In carefully examining the vascular distribution associated with DCIS, Guidi et al. found they could be categorized into two patterns: “diffusely” permeating blood vessels in the stroma, or vascular “cuffing” wherein blood vessels pack densely around neoplastic ducts (Guidi et al. 1994). Expression of VEGF, a potent angiogenic factor, has been found in approximately 85 % of DCIS lesions (Hieken et al. 2001).
Other markers. Other important molecular markers including cell-cycle regulators, proliferation, and apoptosis markers have been found to be differentially expressed in subsets of DCIS. These include cyclin A, cyclin E, p21, p27, Bcl-2, Bax, Survivin, Ki-67, and COX-2 (Clark et al. 2011; Lari and Kuerer 2011). The latter two, used often to characterize invasive carcinomas, may be particularly relevant for DCIS. Ki-67 is a nuclear protein used as a cellular marker for proliferation. Approximately 40 % of DCIS lesions are positive for Ki-67 (Livasy et al. 2007; Kerlikowske et al. 2010). Increased Ki-67 expression is associated with high grade and comedo necrosis (Lari and Kuerer 2011). Alterations to COX-2 expression and the accumulation of its enzymatic product prostaglandins have been implicated in a variety of human cancers, including approximately 45–65 % of DCIS (Boland et al. 2004; Kerlikowske et al. 2010; Glover et al. 2011). COX-2 expression has been associated with high nuclear grade and the Ki-67+/ER− molecular signatures (Lari and Kuerer 2011).
In analyzing protein expression levels, we have identified key pathways governing proliferation, apoptosis, and genomic stability that are altered in DCIS, and have also revealed subtypes of DCIS wherein molecular markers are differentially expressed i.e., exhibit marked inter-tumor heterogeneity. However, DCIS also exhibits considerable intra-tumor heterogeneity as it is often possible to find a variety of grades and molecular subtypes within the same lesion (Polyak 2007). This heterogeneity could be explained by several possible mechanisms, including genomic instability due to telomere shortening, epigenetic instability, malignant stem cells, or phenotypic plasticity (Chin et al. 2004; Marusyk and Polyak 2010). In studying clonal diversity, Allred et al. observed that 50 % of DCIS lesions include diverse nuclear grades and molecular markers, and that this intratumor heterogeneity correlated with p53 expression on IHC (Allred et al. 2008). They proposed that aberrant p53 activity leads to genetic instability and the development of multiple neoplastic clones within the same lesion. These diverse regions in DCIS then compete for dominance, suggesting that poorly differentiated DCIS evolves from well-differentiated DCIS by randomly acquiring genetic defects. Intratumor heterogeneity itself may be a useful molecular marker for the potential of preinvasive cancers to progress to invasion—clonal diversity measures in Barret’s esophagus can predict progression to esophageal adenocarcinoma (Maley et al. 2006)—although this has not been fully explored in DCIS. The mechanisms underlying the etiology of intratumor heterogeneity in DCIS are still poorly defined. They may be better understood within the context of models of preinvasive cancer progression.
3 Progression of DCIS to Invasive Disease
Until now we have mostly focused on a thorough molecular characterization of DCIS at the snapshot in time that it was diagnosed. But how does DCIS evolve over time and progress to invasive carcinoma? Investigating this question will not only shed light on the etiology of breast cancer, but will also address the critical clinical goal of identifying subsets of DCIS patients based on risk of progression to invasive carcinoma (Allegra et al. 2010). With this clinical motivation in mind, we will ask the following two questions of the DCIS progression models we present below: given a newly diagnosed DCIS lesion, can it (i) predict the type of invasive cancer that will arise? and (ii) determine the risk of progression to invasive cancer?
3.1 Predictive Markers of DCIS Progression
The empirical data regarding molecular attributes that can predict risk of DCIS progression to IDC are murky due to the tremendous challenge of performing such investigations in humans. Limitations include small population sizes, lack of long-term follow-up of outcome, and difficulty controlling for different treatments. Perhaps most constraining is the clinical necessity of surgical excision of DCIS instead of observation—this precludes following its natural history to truly determine which features of DCIS predict the risk of progression to invasive carcinoma. As a surrogate endpoint, recurrence is used to assess progression of disease in most studies (Kerlikowske et al. 2010; Lari and Kuerer 2011). Higher nuclear grade, comedo growth pattern, necrosis, and positive surgical margins are linked with increased rates of local recurrence in DCIS (Shamliyan et al. 2010). Additionally, there is some evidence implicating the following molecular markers as correlating with a subsequent recurrence event (Table 1): ER−, PR−, HER2+, Ki67+, cyclin D1−, p21+, p53+, BCL-2−, Survivin+ and possibly most convincingly, p16+ and COX-2+ (Lari and Kuerer 2011; Rakovitch et al. 2012). The largest study to date was performed by Kerlikowske et al. examining a diversity of molecular markers in 1162 women diagnosed with DCIS (Kerlikowske et al. 2010). They found that the molecular signature p16+/COX-2-/Ki-67+ was predictive of a subsequent DCIS but not invasive recurrence, while the p16+/COX-2+/Ki-67+ signature was predictive of a subsequent invasive recurrence. This establishes a role for COX-2 in promoting invasive potential of DCIS, and also implicates aberrant Rb pathway activity in the p16+/Ki-67+ signature (Gauthier et al. 2007; Witkiewicz et al. 2011). They also found the ER−/HER2+ and ER−/HER2+/Ki-67+ signatures to correlate with DCIS recurrence. Although there are many caveats in such studies, the empirical data suggests it is possible for distinct molecular signatures to predict risk of DCIS progression to invasive carcinoma. But is this possibility consistent with models of breast cancer evolution?
3.2 Models of DCIS Progression
The traditional sequential model of cancer progression that has been proposed for many tissue types (Vogelstein et al. 1988) posits that genetic and epigenetic modifications accumulate gradually over time, governing the transition from normal to in situ and then invasive carcinoma (Fig. 2). Consequently, at the time of DCIS diagnosis it would not be possible to predict either the risk of progression or the type of invasive cancer that could arise because the transforming biological events conferring invasion have likely not yet occurred. However, the similarities we have seen between DCIS and IDC on the genome and transcriptome level leaves little room for genetic events to dictate the transition from preinvasive to invasive disease. Rather, it suggests that the bulk of malignant transformation within the epithelial cells has already occurred by the DCIS stage.
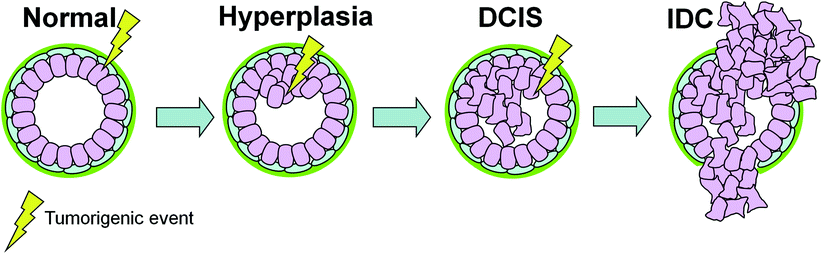
Fig. 2
The sequential model of DCIS progression. Tumorigenic events (e.g., oncogene activation, loss of tumor suppressor) targeting epithelial cells govern the transition between the different stages of breast cancer. In this model, an additional genetic hit is required for progression from DCIS to IDC. Therefore, at the time of DCIS diagnosis it is not possible to predict the type of invasive cancer that will arise
One possible mechanism for this relates to the cancer stem cell hypothesis (Fig. 3). As suggested by Cardiff et al. and others, a precancer stem cell, capable of self-renewal and reestablishing a preinvasive cancer, could be the cell of origin for DCIS (Cardiff and Borowsky 2010). An initiating tumorigenic event occurring in a precancer stem cell would in turn generate an intrinsic subtype of cancer based on both the characteristics of the initiating event itself and the target precancer stem cell. The biology of the lesion including its potential for progression to IDC is thus ‘pre-encoded’ in its preinvasive state. In this way, the critical somatic mutations that determine the cell’s fate have already occurred before the appearance of DCIS and do not change significantly with progression to IDC. Subsequent epigenetic or microenvironment effects can modulate the timing of progression (Cardiff and Borowsky 2010).