Interpretation of Copy Number Alterations Identified Through Clinical Microarray-Comparative Genomic Hybridization
Keywords
• Oligonucleotide microarray • Copy number alteration • Variant of unknown significance • Deletion • Duplication
Array-comparative genomic hybridization (array-CGH) has become the first-tier technology for the clinical assessment of copy number alterations (CNAs) associated with features such as developmental delay, congenital malformations, autism, or dysmorphic features. It is a near certainty that at least 1 CNA will be identified in each array-CGH assay and the challenge then becomes differentiating between benign CNA and those copy number changes that are dosage sensitive and relate to the abnormal phenotype in question. With advances in array-CGH platform coverage, it is no longer a question of CNA identification, but rather of CNA interpretation. However, there is no single methodology employed by laboratories to conduct this process, and unfortunately all too often the end results provide no clear answer to the ordering physician or their patients. Recent approaches for interpretation of CNA have advanced to suggesting the use of first-generation computational models based on structural and functional genomic features to predict a pathologic or benign value for array-CGH findings.1 Although such estimation procedures can be helpful, they are currently limited to a single phenotype (mental retardation) and can at best only provide an educated guess as to the true clinical significance of these alterations.
Classification Strategy
A majority of clinical laboratories employ a 3-tiered system for the classification and reporting of array-CGH CNA: Likely pathogenic/abnormal, likely benign (LB), and variant of unknown significance (VUS).2–4 For a given CNA, this process typically involves assessing the size, content, associations with benign CNA and abnormal phenotypes, and mode of inheritance, if possible. In general, larger abnormalities are thought to be more significant than smaller ones, and duplications are thought to be better tolerated than deletions. Greater significance is placed on alterations in gene-rich regions; however, this interpretation is tempered by correlation with regions of reportedly benign CNA. Large population studies documenting the instances of CNA in normal populations such as those collected within the Database of Genomic Variants5 are extremely important to consult when beginning to identify CNA with benign features. Additionally, many laboratories maintain their own databases of benign CNA as another level of internal assessment. Connections of CNA to pathogenic features can be examined through online databases such as DECIPHER,6 which collects information on submicroscopic chromosome imbalances, the Online Mendelian Inheritance in Man (OMIM),7 which collects information on human genes, function, and related disorders, and, most important, through the published literature of related cases. Finally, assessing the mode of inheritance for a CNA can suggest a more benign character if the abnormality was inherited from a normal parent or a more potentially significant one if it has occurred de novo.
As recently as 3 years ago, a majority of laboratories were not typically including reportedly benign CNA along with likely pathogenic/abnormal, LB, and VUS findings on their clinical reports because there was thought to be no connection between these genomic alterations and abnormal phenotypes.8 Many laboratories have reconsidered this practice as information has arisen suggesting that some CNA previously thought to be benign may instead have pathogenic implications. For example, microdeletions at chromosome 15q11.2 involving 4 genes, TUBGCP5, NIPA2, NIPA1, and CYFIP1, were initially thought to be benign but recently have been shown to segregate in families with features including learning delay, delayed development, and behavior issues.9 The inclusion of all identified benign CNA in a clinical report could serve as a record of all noted array-CGH abnormalities in case such re-interpretation is ever needed in the future, but would also present a considerable burden for those responsible for writing the report.
Many laboratories utilize a size threshold, from 50 to 500 kb for deletions and 150 to 500 kb for duplications, to define what array-CGH abnormalities will or will not be clinically reported.2 This approach can help to reduce the number of CNA of unknown significance requiring investigation, but additionally can erroneously exclude clinically significant CNA of small size. As an alternative approach to size exclusion, laboratories can utilize an approach based on the number of oligonucleotide probes altered and the significance of the shift from copy number neutral value. Although there is currently no consensus regarding the use of size cutoffs, the American College of Medical Genetics recommendations10 for the clinical use of array-CGH may provide some guidance.
VUS
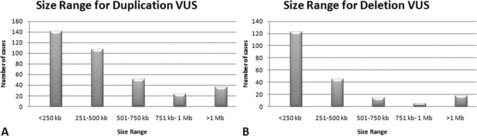
Regardless of the interpretation strategy used, VUS are a large portion of array-CGH findings and require a significant amount of time to investigate. From a consecutive series of 1998 cases submitted to our laboratory for clinical array-CGH analysis using a custom, 105,000 oligonucleotide array platform, 563 abnormalities were interpreted as a VUS from 490 patients (24.5% of cases). The 358 duplication VUS from this series ranged in size from 33 kb to 2.9 Mb with a majority being 500 kb or less (Fig. 1A). The 205 deletion VUS ranged in size from 24 kb to 1.54 Mb and were significantly smaller, with the majority being 250 kb or less (see Fig. 1B). Duplications in this series were more frequent than deletions as losses of genetic material are thought to be less well-tolerated than gains.11 These values are similar to previously published frequencies of copy number variants in phenotypically normal individuals (65%–80% 100–500 kb; 5%–10% >500 kb; 1% >1 Mb12), which suggests a majority of these findings may be similarly benign.
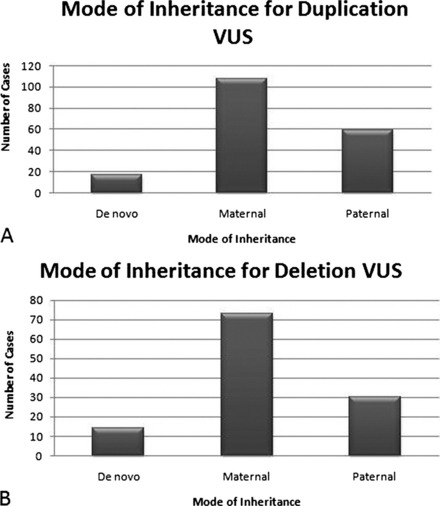
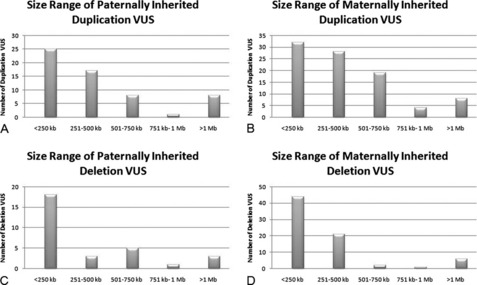
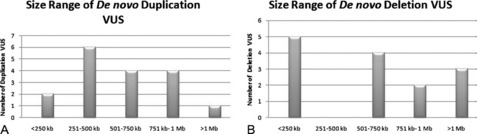
Parental samples should always be requested for VUS to help define the significance of the abnormality. In a practice adopted from conventional cytogenetics, copy number changes that are found to be de novo are thought to be more likely pathogenic. However, in these cases, false paternity must be considered when counseling the significance of such a finding. The demonstration of CNA inheritance from a phenotypically normal parent suggests a more benign character to the alteration. However, if the normal phenotype in the parent is in question or cannot be firmly established, additional pedigree analysis may be warranted to track the occurrence of the phenotype and inheritance of the CNA within the family. Of the 285 cases from our laboratory in which the mode of inheritance for the VUS could be determined, a majority of both duplication and deletion VUS were maternally inherited (Fig. 2A, B). Of 167 duplication VUS, 91 (54.5%) were maternally inherited, 59 (35.3%) were paternal, and 17 (10.2%) were determined to be de novo in origin (see Fig. 2A). Similar proportions were observed for deletion VUS with 74 (62.7%) maternal, 30 (25.4%) paternal, and 124 (11.9%) de novo (see Fig. 2B). The size distribution for both maternally and paternally inherited VUS were similar to the entire respective series of duplication or deletion VUS with a majority of inherited duplication VUS being 500 kb or less and inherited deletion VUS being less than 250 kb (Fig. 3). No such trend was observed for de novo VUS, with sizes of 500 kb or greater being more common for both deletions and duplications (Fig. 4). Based on these results, parental analysis of VUS in our laboratory is conducted in a sequential manner starting with maternal samples, because this method will identify the inheritance in more than half of the cases while requiring the analysis of only a single parental sample.
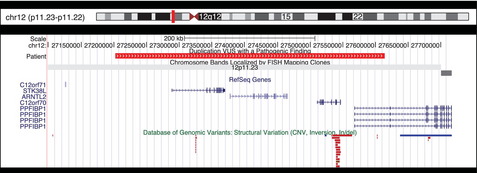
In approximately 60% of cases in this series, the deletion or duplication VUS was the sole abnormality identified. In 34 cases, deletion or duplication VUS were co-identified along with a pathogenic finding. These instances can provide additional insight into the significance of variants because they represent either the inheritance of 2 pathogenic alterations in the same individual, the co-inheritance of a modifying factor for the pathogenic alteration, or the incidental inheritance of a benign variant with the pathogenic one. Fig. 5 shows one such VUS. This approximately 411kb gain at chromosome 12p11.23 includes the coding regions of 2 genes (STK38L and ARNTL2), 2 open reading frames (C12orf71 and C12orf70), and a portion of a third gene (PPFIBP1). This duplication has been observed as the sole finding in 2 cases, both times being paternally inherited, and twice in conjunction with other variants of unknown significance. In 2 additional cases, this VUS was co-identified with a pathogenic finding including a 1.24Mb terminal loss from 22q13.33 and a 5.9Mb de novo gain at 9q21.2 through 9q13.32. In this latter case, the 12p11.23 variant was determined to have been inherited from a reportedly phenotypically normal mother. Although modifying effects cannot be excluded, the patient’s phenotype seems most likely attributed to the large, de novo duplication on chromosome 9, suggesting benign consequences for the 12p11.23 gain.
Alteration Size Versus Content
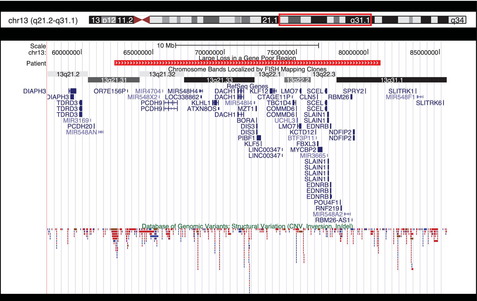
Segmental aneuploidy is often thought to be clinically significant, especially in cases involving alterations which are cytogenetically visible. Fig. 6 shows an 18.55-Mb deletion at chromosome 13q21.31 through 13q31.1 identified in our laboratory. The large size of this loss suggests it is a pathogenic alteration; however, the region is gene poor, containing only 31 genes and 4 microRNAs. Alterations on the long arm of chromosome 13 are typically terminal losses, which are more distal to the region deleted here, and are associated with features including mental/growth retardation, dysmorphic features, malformations, holoprosencephaly, Dandy–Walker malformation, and digit abnormalities.13–16 The literature contains 2 reports of comparable alterations at this interstitial 13q region and the associated features paint a different picture. These include a 11.2-Mb duplication at 13q12, which overlaps the proximal portion of the deleted region in Fig. 6, including 2 genes (OR7E1569 and PCDH9) in a boy with autism17 and a 14.5-Mb deletion identified prenatally in a male child who presented with hypotonia at birth and later with some pigmentation anomalies and slightly delayed motor development. This deletion was show to have been inherited from the child’s mother and maternal grandfather, both of whom were phenotypically normal.18 Finally, a 29.8-Mb deletion at 13q21.31 through 13q31.3 was reported in a 16-month-old girl who demonstrated micrognathia, tapering fingers, mild dysmorphic features, hypotonia, short stature, microcephaly, and moderate mental retardation.19 Using a series of cases in addition to that one, the authors assembled a genotype–phenotype map for deletions on the long arm of chromosome 13 with micrognathia being the only feature associated with losses in the 13q21.32 through 13q31.1 region. Features such as Dandy–Walker malformations, microcephaly, and limb abnormalities were associated with more distal critical segments on 13q.19 Based on these findings, including a lack of association with major features, demonstration of stable, multigenerational inheritance from normal individuals, and the gene-poor content of this region, alterations in this region have been proposed as benign variants.17,18 However, additional studies are needed to firmly establish the benign nature of these variants on chromosome 13 and the 18.55-Mb deletion observed in our laboratory was consequently reported as a VUS.
Related posts:
 Cytogenetics
Cytogenetics
 Structural Rearrangements: Detection and Elucidation of Mechanisms Using Cytogenomic Technologies
Structural Rearrangements: Detection and Elucidation of Mechanisms Using Cytogenomic Technologies
 Evolving Picture of Microdeletion/Microduplication Syndromes in the Age of Microarray Analysis: Variable Expressivity and Genomic Complexity
Evolving Picture of Microdeletion/Microduplication Syndromes in the Age of Microarray Analysis: Variable Expressivity and Genomic Complexity
 In Situ Hybridization
In Situ Hybridization
 Myeloma: Current Perspectives
Myeloma: Current Perspectives
 Syndromes
Syndromes
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


